

Multi-task bioassay pre-training for protein-ligand binding affinity prediction
- PMID: 38084920
- PMCID: PMC10783875
- DOI: 10.1093/bib/bbad451
Multi-task bioassay pre-training for protein-ligand binding affinity prediction
Abstract
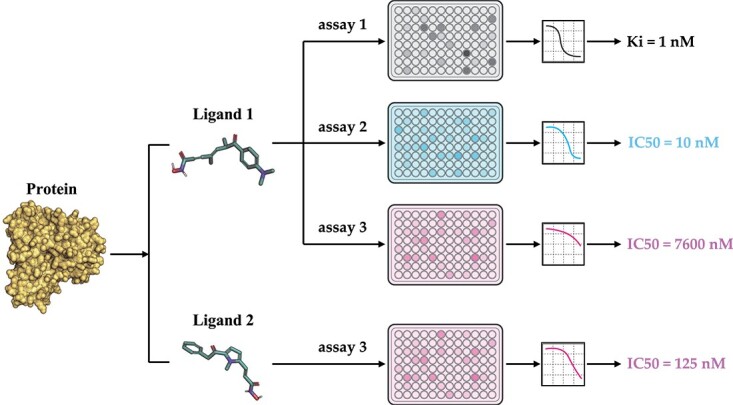
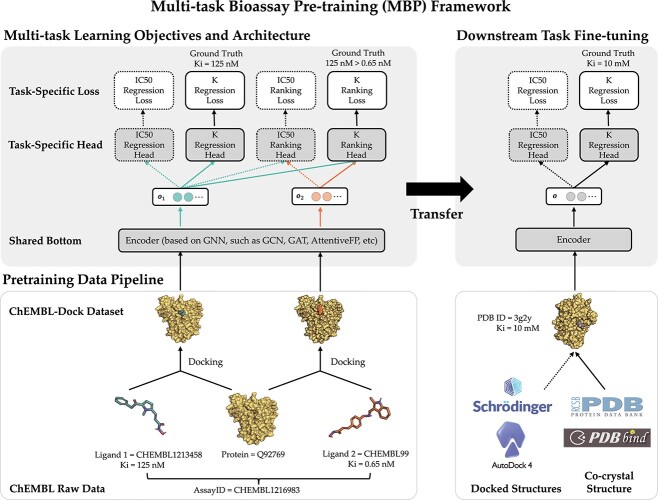
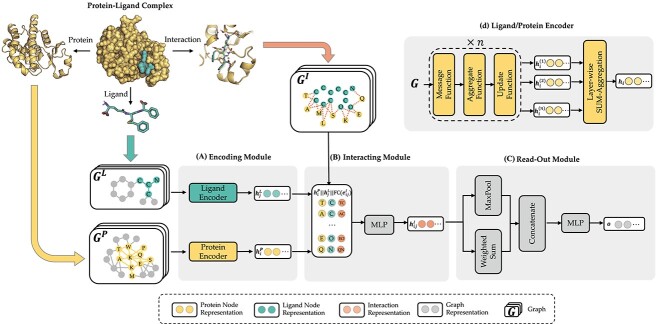
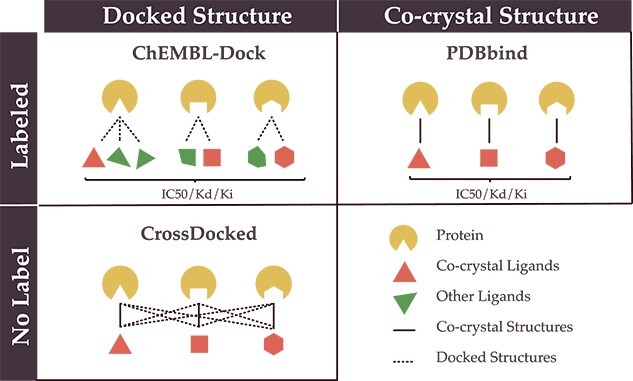
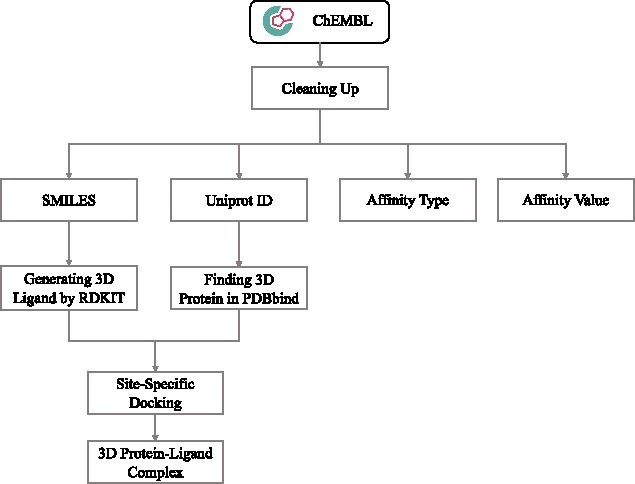
Protein-ligand binding affinity (PLBA) prediction is the fundamental task in drug discovery. Recently, various deep learning-based models predict binding affinity by incorporating the three-dimensional (3D) structure of protein-ligand complexes as input and achieving astounding progress. However, due to the scarcity of high-quality training data, the generalization ability of current models is still limited. Although there is a vast amount of affinity data available in large-scale databases such as ChEMBL, issues such as inconsistent affinity measurement labels (i.e. IC50, Ki, Kd), different experimental conditions, and the lack of available 3D binding structures complicate the development of high-precision affinity prediction models using these data. To address these issues, we (i) propose Multi-task Bioassay Pre-training (MBP), a pre-training framework for structure-based PLBA prediction; (ii) construct a pre-training dataset called ChEMBL-Dock with more than 300k experimentally measured affinity labels and about 2.8M docked 3D structures. By introducing multi-task pre-training to treat the prediction of different affinity labels as different tasks and classifying relative rankings between samples from the same bioassay, MBP learns robust and transferrable structural knowledge from our new ChEMBL-Dock dataset with varied and noisy labels. Experiments substantiate the capability of MBP on the structure-based PLBA prediction task. To the best of our knowledge, MBP is the first affinity pre-training model and shows great potential for future development. MBP web-server is now available for free at: https://huggingface.co/spaces/jiaxianustc/mbp.
Keywords: bioassay; graph neural network; pre-training; protein–ligand binding affinity.
© The Author(s) 2023. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
GeneralizedDTA: combining pre-training and multi-task learning to predict drug-target binding affinity for unknown drug discovery.BMC Bioinformatics. 2022 Sep 7;23(1):367. doi: 10.1186/s12859-022-04905-6. BMC Bioinformatics. 2022. PMID: 36071406 Free PMC article.
-
ERL-ProLiGraph: Enhanced representation learning on protein-ligand graph structured data for binding affinity prediction.Mol Inform. 2024 Dec;43(12):e202400044. doi: 10.1002/minf.202400044. Epub 2024 Oct 15. Mol Inform. 2024. PMID: 39404190 Free PMC article.
-
PLANET: A Multi-objective Graph Neural Network Model for Protein-Ligand Binding Affinity Prediction.J Chem Inf Model. 2024 Apr 8;64(7):2205-2220. doi: 10.1021/acs.jcim.3c00253. Epub 2023 Jun 15. J Chem Inf Model. 2024. PMID: 37319418
-
Learning from Docked Ligands: Ligand-Based Features Rescue Structure-Based Scoring Functions When Trained on Docked Poses.J Chem Inf Model. 2022 Nov 28;62(22):5329-5341. doi: 10.1021/acs.jcim.1c00096. Epub 2021 Sep 1. J Chem Inf Model. 2022. PMID: 34469150 Review.
-
[Advances in using artificial intelligence for predicting protein-ligand binding affinity].Sheng Wu Gong Cheng Xue Bao. 2024 Jul 25;40(7):2070-2086. doi: 10.13345/j.cjb.230679. Sheng Wu Gong Cheng Xue Bao. 2024. PMID: 39044576 Review. Chinese.
Cited by
-
EM-PLA: environment-aware heterogeneous graph-based multimodal protein-ligand binding affinity prediction.Bioinformatics. 2025 Jul 1;41(7):btaf298. doi: 10.1093/bioinformatics/btaf298. Bioinformatics. 2025. PMID: 40354612 Free PMC article.
-
Predicting Affinity Through Homology (PATH): Interpretable binding affinity prediction with persistent homology.PLoS Comput Biol. 2025 Jun 27;21(6):e1013216. doi: 10.1371/journal.pcbi.1013216. eCollection 2025 Jun. PLoS Comput Biol. 2025. PMID: 40577377 Free PMC article.
-
Assay2Mol: Large Language Model-based Drug Design Using BioAssay Context.ArXiv [Preprint]. 2025 Jul 16:arXiv:2507.12574v1. ArXiv. 2025. PMID: 40709303 Free PMC article. Preprint.
References
-
- Rizzuti B, Grande F. Chapter 14- virtual screening in drug discovery: a precious tool for a still-demanding challenge. In: Pey AL (ed). Protein Homeostasis Diseases. Academic Press, United States, 2020, 309–27.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
