

Single-cell spatial metabolomics with cell-type specific protein profiling for tissue systems biology
- PMID: 38086839
- PMCID: PMC10716522
- DOI: 10.1038/s41467-023-43917-5
Single-cell spatial metabolomics with cell-type specific protein profiling for tissue systems biology
Abstract
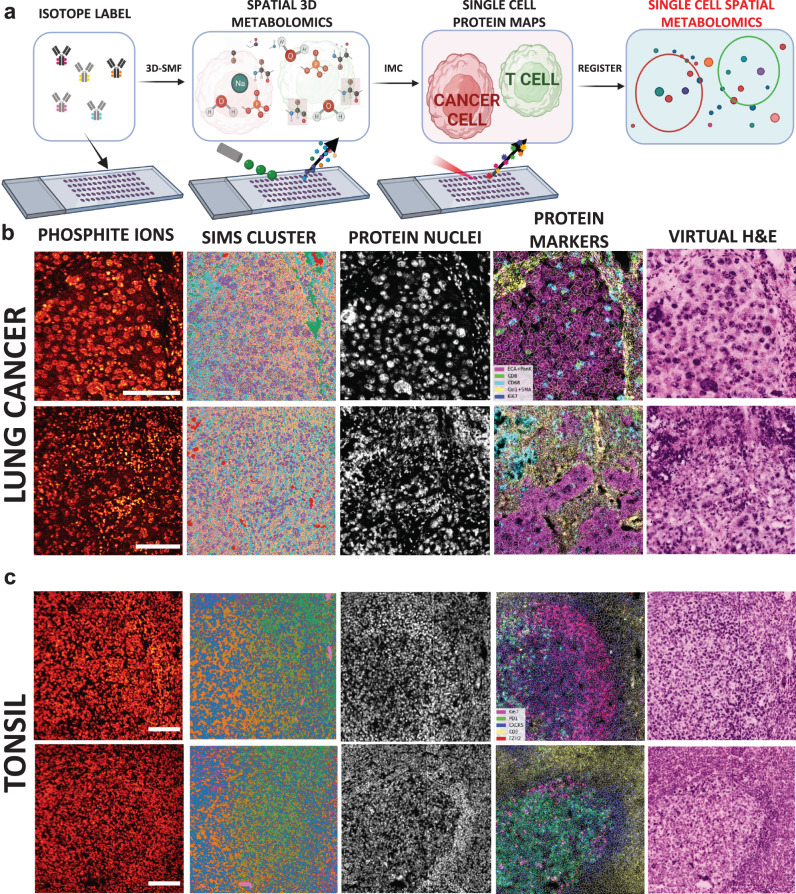
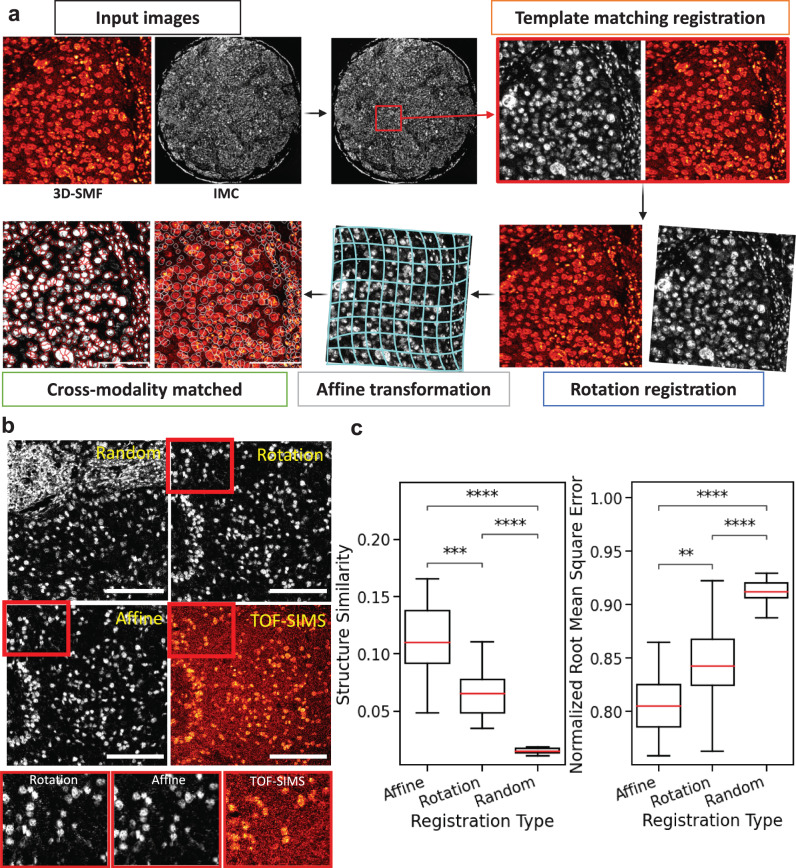
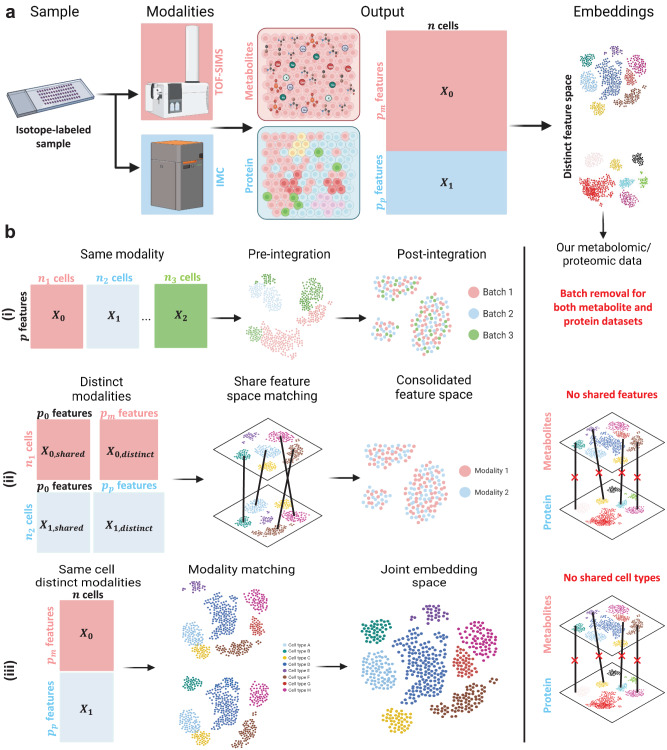
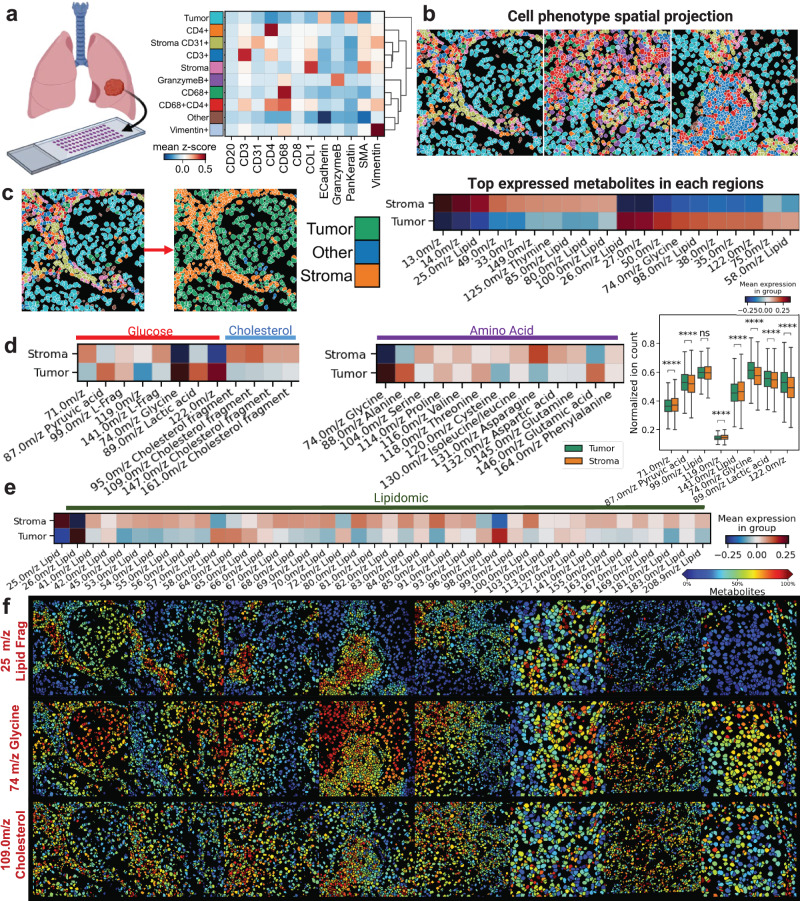
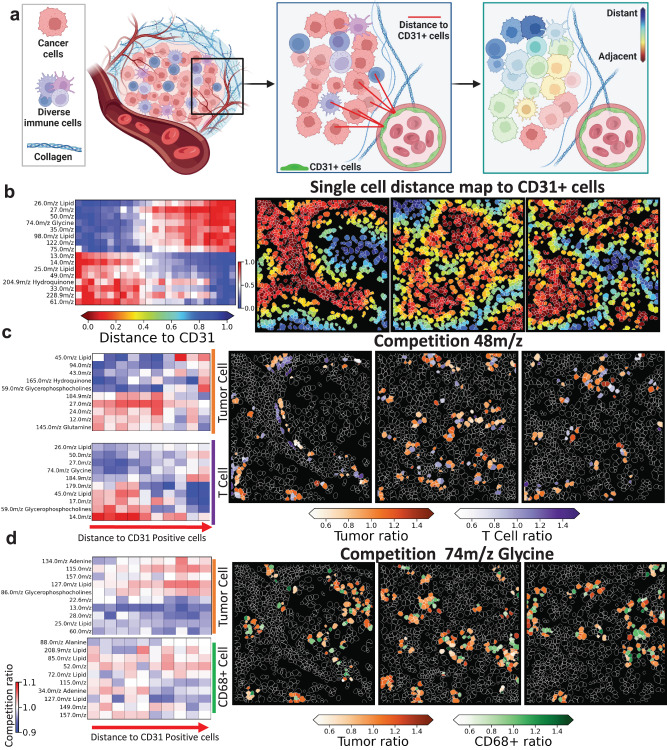
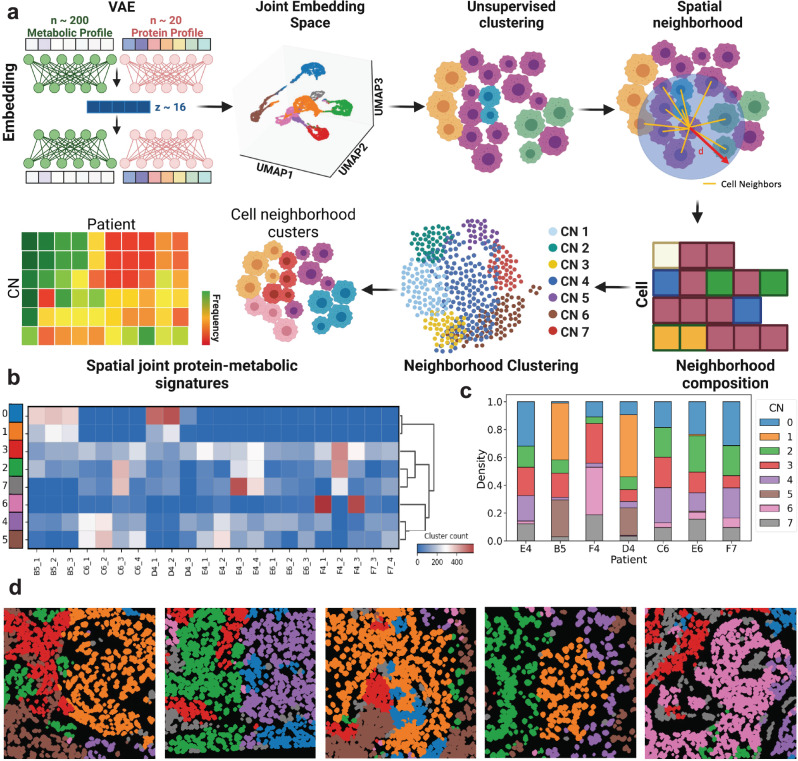
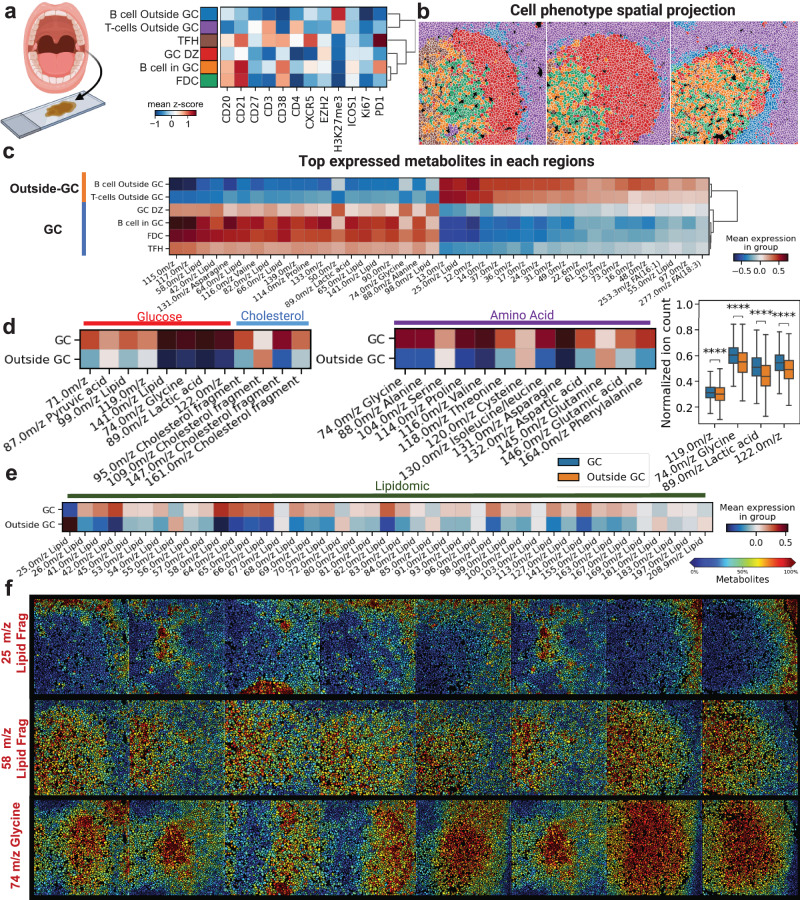
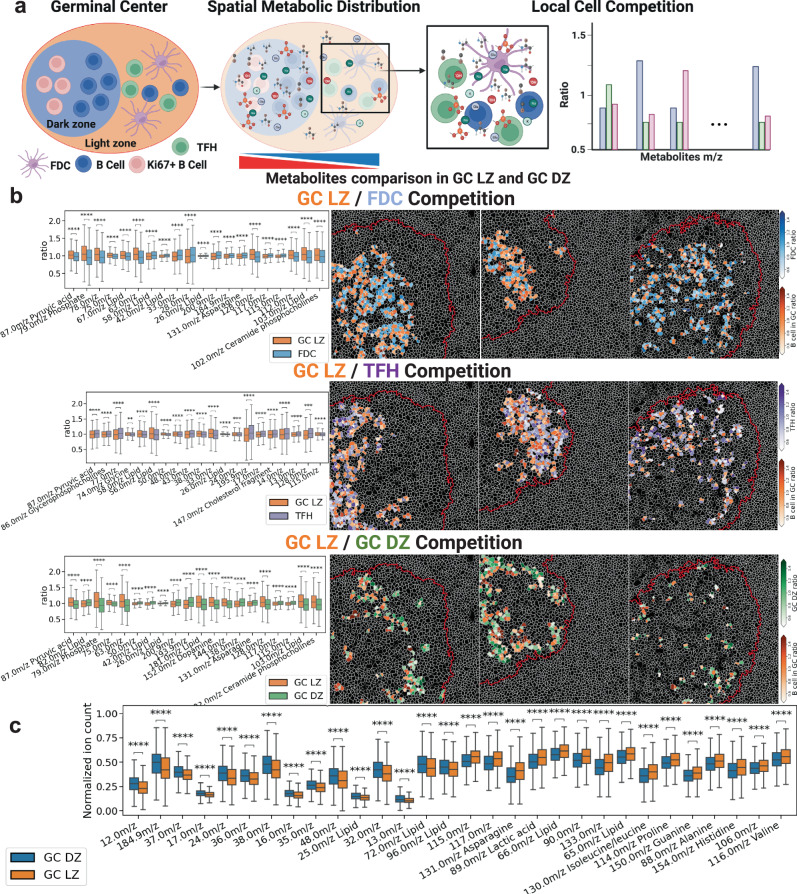
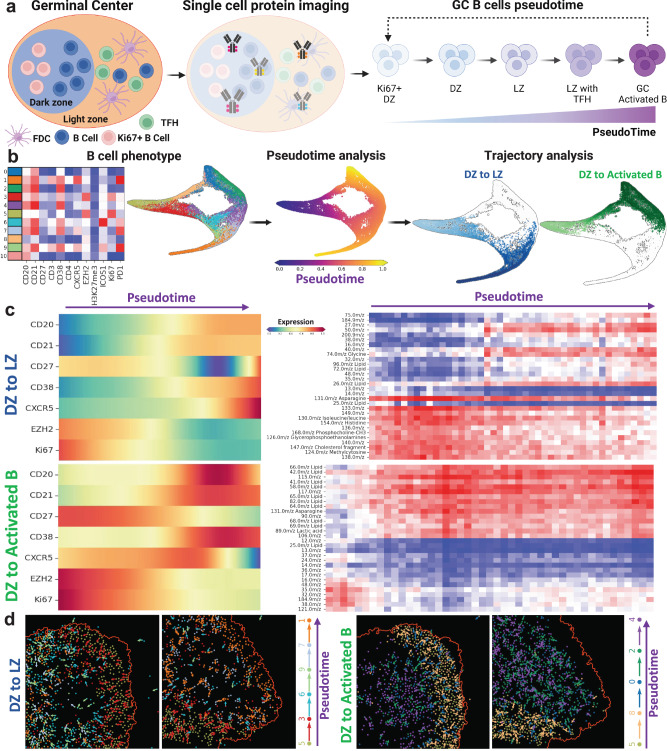
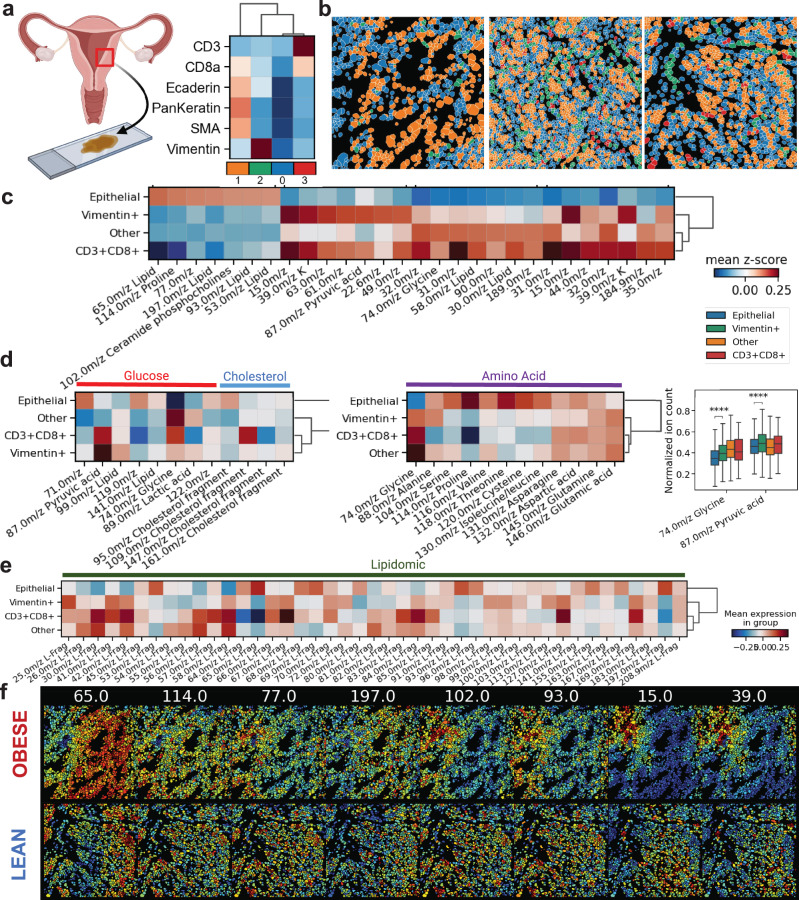
Metabolic reprogramming in cancer and immune cells occurs to support their increasing energy needs in biological tissues. Here we propose Single Cell Spatially resolved Metabolic (scSpaMet) framework for joint protein-metabolite profiling of single immune and cancer cells in male human tissues by incorporating untargeted spatial metabolomics and targeted multiplexed protein imaging in a single pipeline. We utilized the scSpaMet to profile cell types and spatial metabolomic maps of 19507, 31156, and 8215 single cells in human lung cancer, tonsil, and endometrium tissues, respectively. The scSpaMet analysis revealed cell type-dependent metabolite profiles and local metabolite competition of neighboring single cells in human tissues. Deep learning-based joint embedding revealed unique metabolite states within cell types. Trajectory inference showed metabolic patterns along cell differentiation paths. Here we show scSpaMet's ability to quantify and visualize the cell-type specific and spatially resolved metabolic-protein mapping as an emerging tool for systems-level understanding of tissue biology.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
