

Restrictor synergizes with Symplekin and PNUTS to terminate extragenic transcription
- PMID: 38092518
- PMCID: PMC10760643
- DOI: 10.1101/gad.351057.123
Restrictor synergizes with Symplekin and PNUTS to terminate extragenic transcription
Abstract
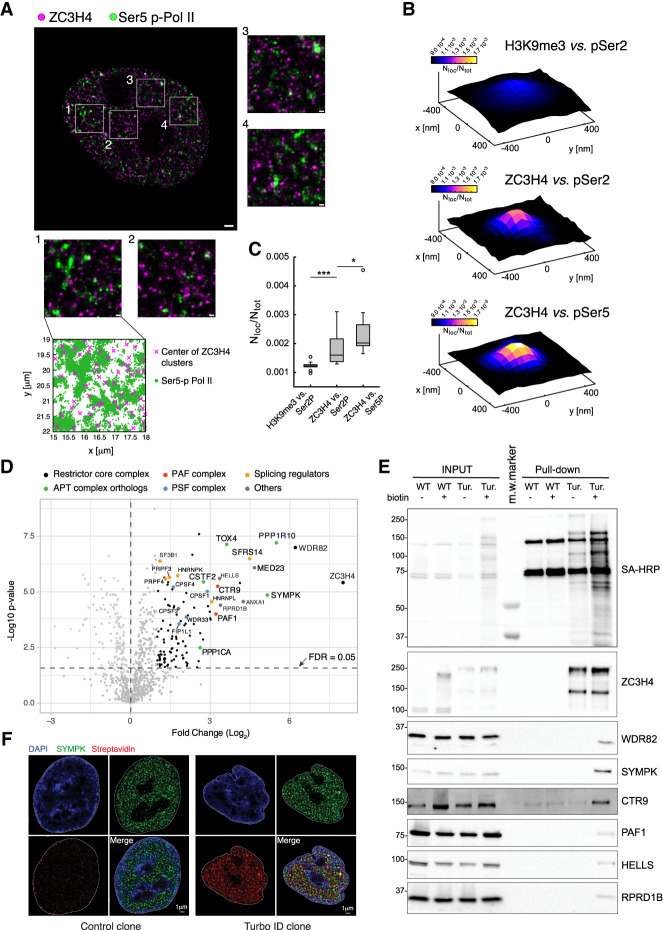
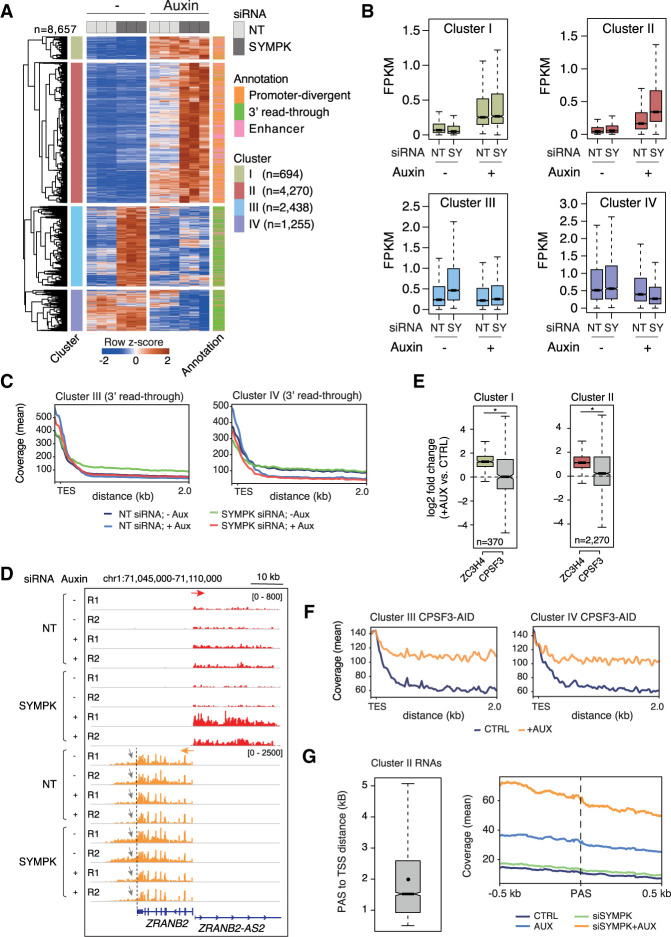
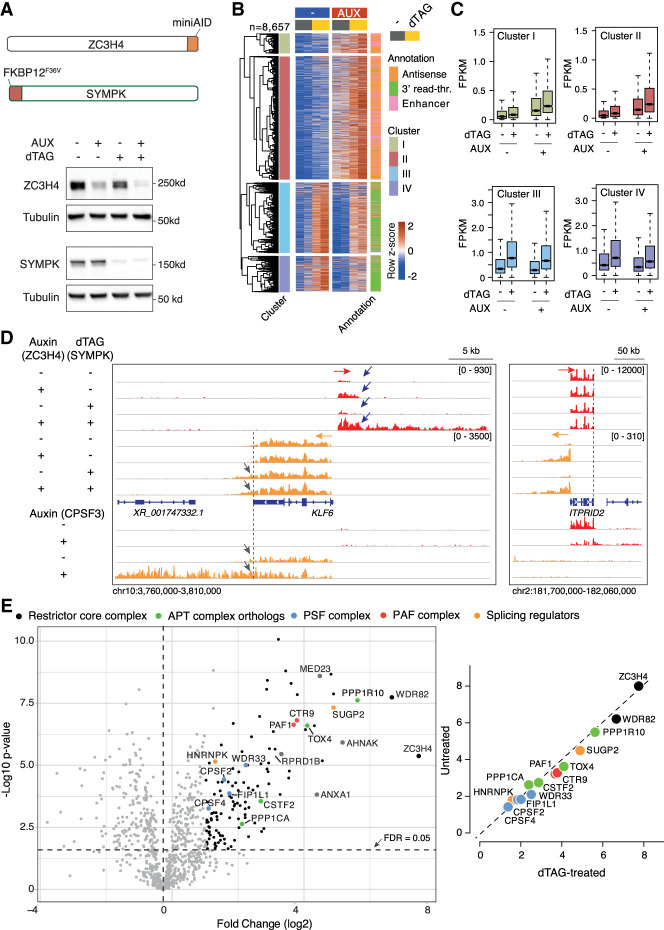
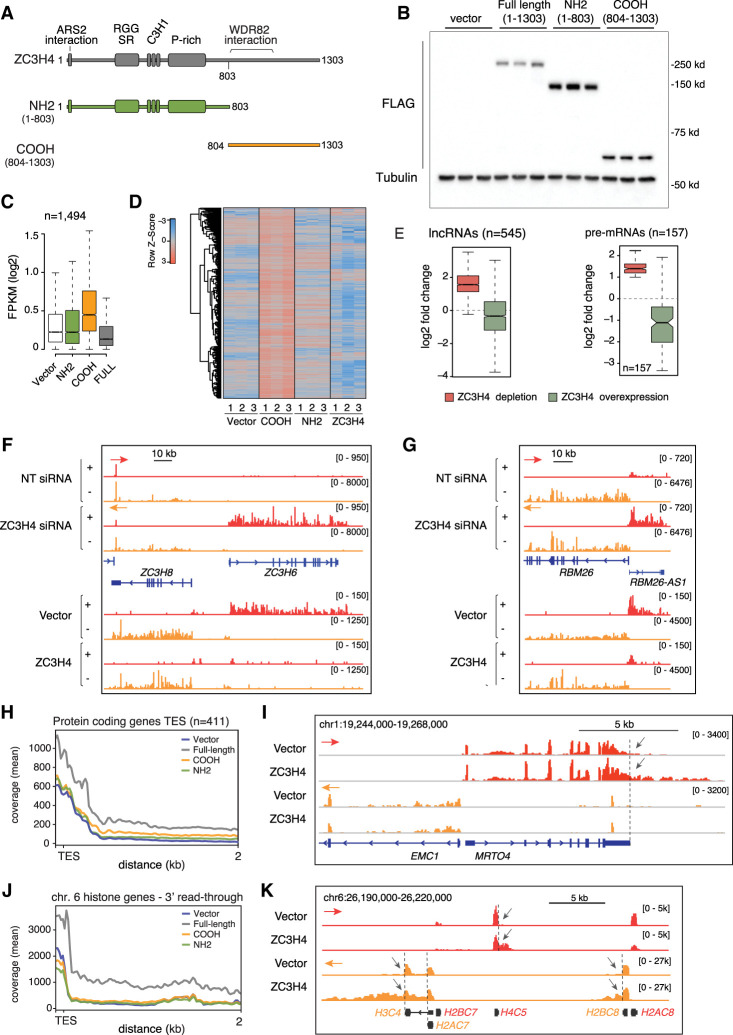
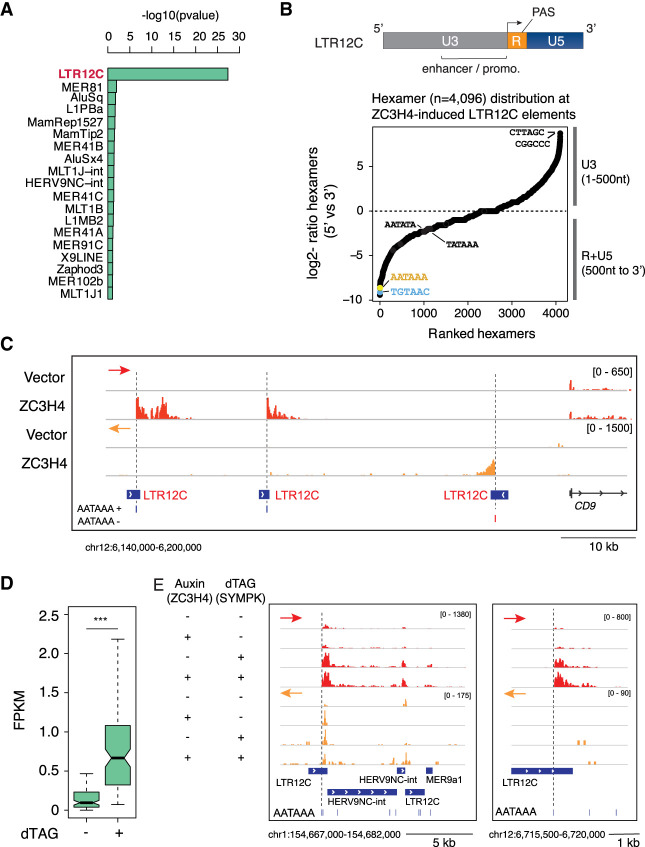
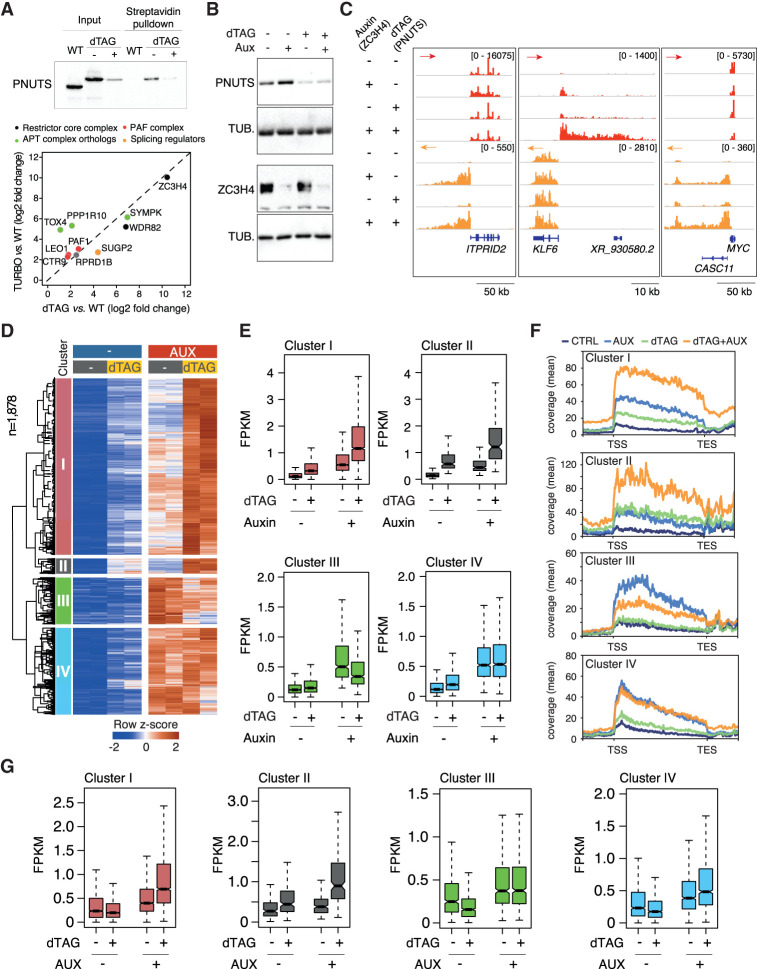
Transcription termination pathways mitigate the detrimental consequences of unscheduled promiscuous initiation occurring at hundreds of thousands of genomic cis-regulatory elements. The Restrictor complex, composed of the Pol II-interacting protein WDR82 and the RNA-binding protein ZC3H4, suppresses processive transcription at thousands of extragenic sites in mammalian genomes. Restrictor-driven termination does not involve nascent RNA cleavage, and its interplay with other termination machineries is unclear. Here we show that efficient termination at Restrictor-controlled extragenic transcription units involves the recruitment of the protein phosphatase 1 (PP1) regulatory subunit PNUTS, a negative regulator of the SPT5 elongation factor, and Symplekin, a protein associated with RNA cleavage complexes but also involved in cleavage-independent and phosphatase-dependent termination of noncoding RNAs in yeast. PNUTS and Symplekin act synergistically with, but independently from, Restrictor to dampen processive extragenic transcription. Moreover, the presence of limiting nuclear levels of Symplekin imposes a competition for its recruitment among multiple transcription termination machineries, resulting in mutual regulatory interactions. Hence, by synergizing with Restrictor, Symplekin and PNUTS enable efficient termination of processive, long-range extragenic transcription.
Keywords: RNA polymerase II; transcription; transcription termination.
© 2023 Russo et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous