

Single-cell DNA methylome and 3D multi-omic atlas of the adult mouse brain
- PMID: 38092913
- PMCID: PMC10719113
- DOI: 10.1038/s41586-023-06805-y
Single-cell DNA methylome and 3D multi-omic atlas of the adult mouse brain
Abstract
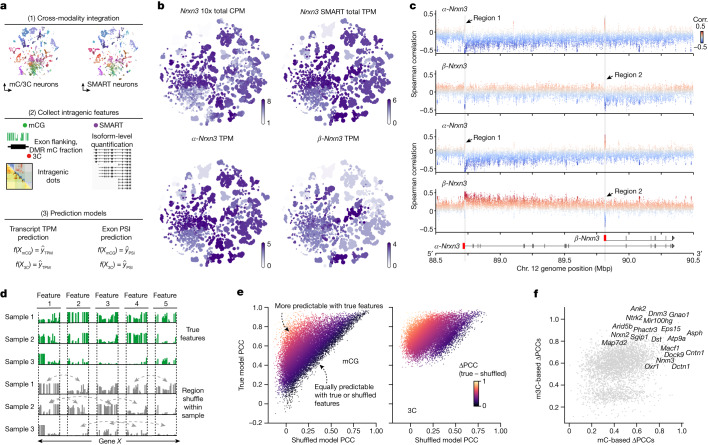
Cytosine DNA methylation is essential in brain development and is implicated in various neurological disorders. Understanding DNA methylation diversity across the entire brain in a spatial context is fundamental for a complete molecular atlas of brain cell types and their gene regulatory landscapes. Here we used single-nucleus methylome sequencing (snmC-seq3) and multi-omic sequencing (snm3C-seq)1 technologies to generate 301,626 methylomes and 176,003 chromatin conformation-methylome joint profiles from 117 dissected regions throughout the adult mouse brain. Using iterative clustering and integrating with companion whole-brain transcriptome and chromatin accessibility datasets, we constructed a methylation-based cell taxonomy with 4,673 cell groups and 274 cross-modality-annotated subclasses. We identified 2.6 million differentially methylated regions across the genome that represent potential gene regulation elements. Notably, we observed spatial cytosine methylation patterns on both genes and regulatory elements in cell types within and across brain regions. Brain-wide spatial transcriptomics data validated the association of spatial epigenetic diversity with transcription and improved the anatomical mapping of our epigenetic datasets. Furthermore, chromatin conformation diversities occurred in important neuronal genes and were highly associated with DNA methylation and transcription changes. Brain-wide cell-type comparisons enabled the construction of regulatory networks that incorporate transcription factors, regulatory elements and their potential downstream gene targets. Finally, intragenic DNA methylation and chromatin conformation patterns predicted alternative gene isoform expression observed in a whole-brain SMART-seq2 dataset. Our study establishes a brain-wide, single-cell DNA methylome and 3D multi-omic atlas and provides a valuable resource for comprehending the cellular-spatial and regulatory genome diversity of the mouse brain.
© 2023. The Author(s).
Conflict of interest statement
J.R.E. serves on the scientific advisory board of Zymo Research. B.R. is a shareholder of Arima Genomics and Epigenome Technologies. H.Z. is on the scientific advisory board of MapLight Therapeutics.
Figures


















Update of
-
Single-cell DNA Methylome and 3D Multi-omic Atlas of the Adult Mouse Brain.bioRxiv [Preprint]. 2023 Apr 18:2023.04.16.536509. doi: 10.1101/2023.04.16.536509. bioRxiv. 2023. Update in: Nature. 2023 Dec;624(7991):366-377. doi: 10.1038/s41586-023-06805-y. PMID: 37131654 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
