

Single-cell analysis of chromatin accessibility in the adult mouse brain
- PMID: 38092917
- PMCID: PMC10719105
- DOI: 10.1038/s41586-023-06824-9
Single-cell analysis of chromatin accessibility in the adult mouse brain
Abstract
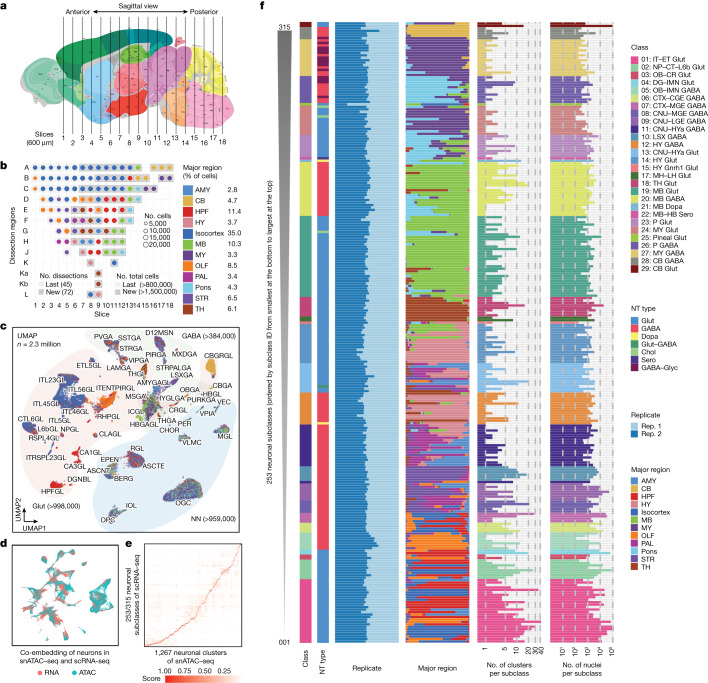
Recent advances in single-cell technologies have led to the discovery of thousands of brain cell types; however, our understanding of the gene regulatory programs in these cell types is far from complete1-4. Here we report a comprehensive atlas of candidate cis-regulatory DNA elements (cCREs) in the adult mouse brain, generated by analysing chromatin accessibility in 2.3 million individual brain cells from 117 anatomical dissections. The atlas includes approximately 1 million cCREs and their chromatin accessibility across 1,482 distinct brain cell populations, adding over 446,000 cCREs to the most recent such annotation in the mouse genome. The mouse brain cCREs are moderately conserved in the human brain. The mouse-specific cCREs-specifically, those identified from a subset of cortical excitatory neurons-are strongly enriched for transposable elements, suggesting a potential role for transposable elements in the emergence of new regulatory programs and neuronal diversity. Finally, we infer the gene regulatory networks in over 260 subclasses of mouse brain cells and develop deep-learning models to predict the activities of gene regulatory elements in different brain cell types from the DNA sequence alone. Our results provide a resource for the analysis of cell-type-specific gene regulation programs in both mouse and human brains.
© 2023. The Author(s).
Conflict of interest statement
B.R. is a co-founder and consultant of Arima Genomics and co-founder of Epigenome Technologies. J.R.E. is on the scientific advisory board of Zymo Research. H.Z. is on the scientific advisory board of MapLight Therapeutics.
Figures





















References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
