

Conserved and divergent gene regulatory programs of the mammalian neocortex
- PMID: 38092918
- PMCID: PMC10719095
- DOI: 10.1038/s41586-023-06819-6
Conserved and divergent gene regulatory programs of the mammalian neocortex
Erratum in
-
Author Correction: Conserved and divergent gene regulatory programs of the mammalian neocortex.Nature. 2024 Jan;625(7996):E26. doi: 10.1038/s41586-023-07013-4. Nature. 2024. PMID: 38200319 Free PMC article. No abstract available.
Abstract
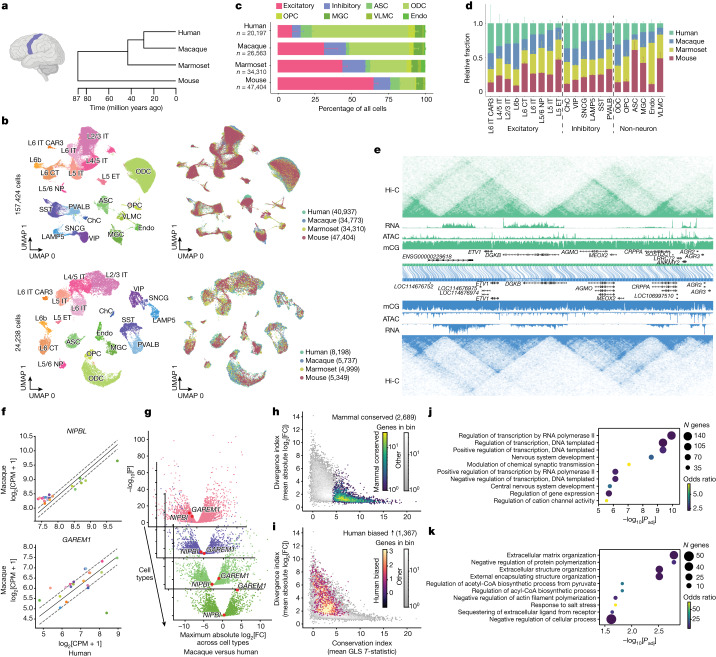
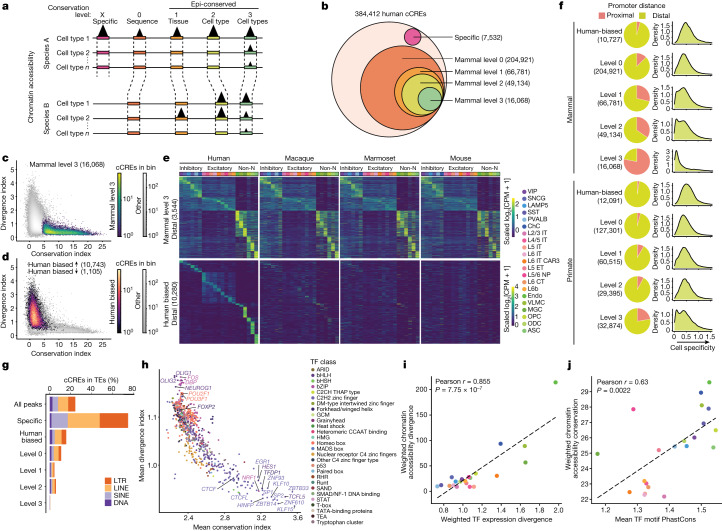
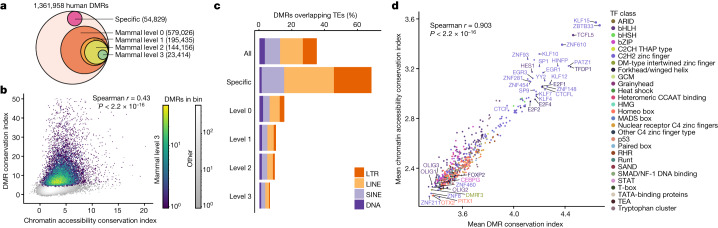
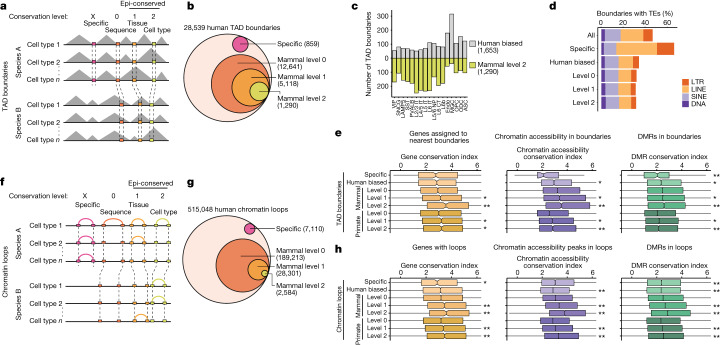
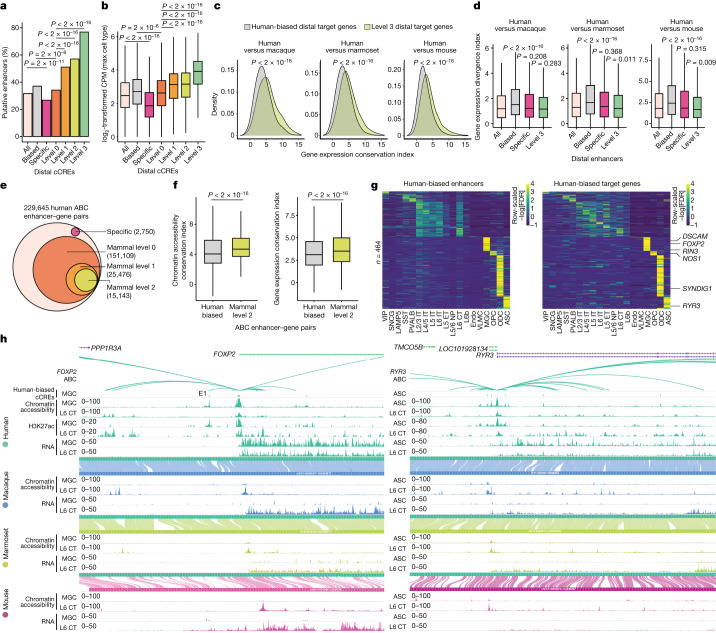
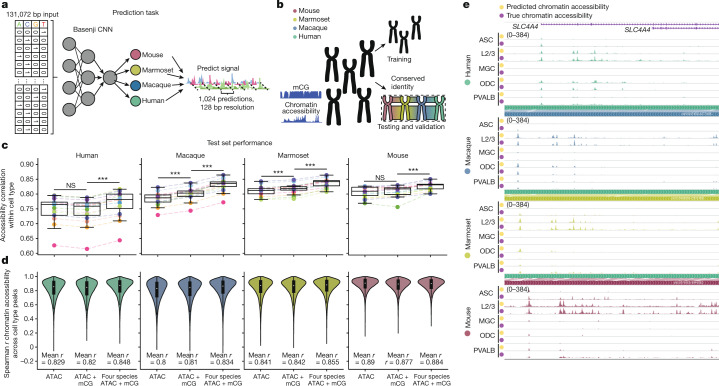
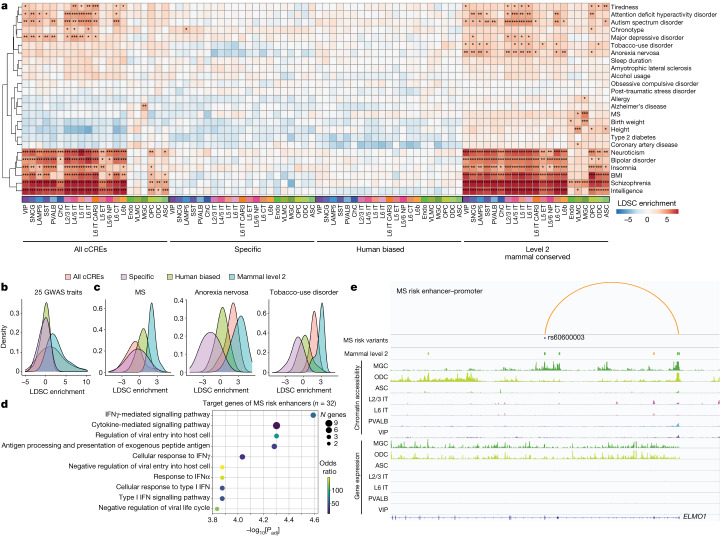
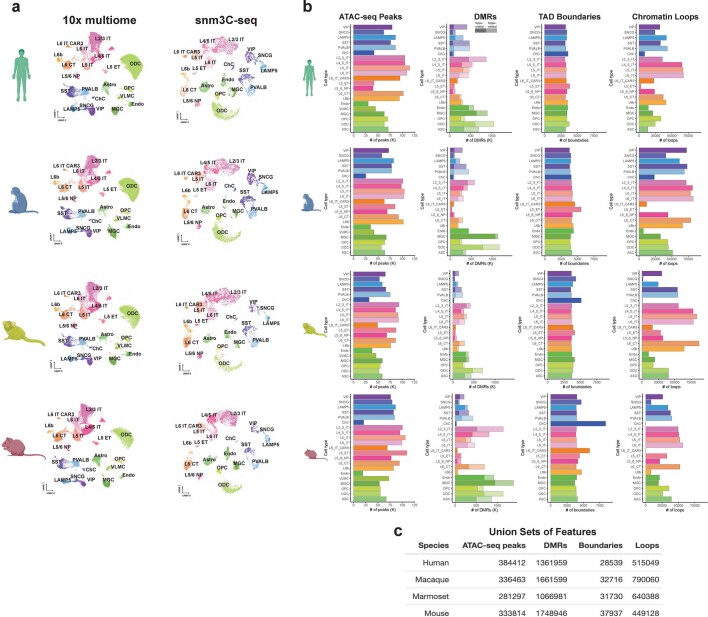
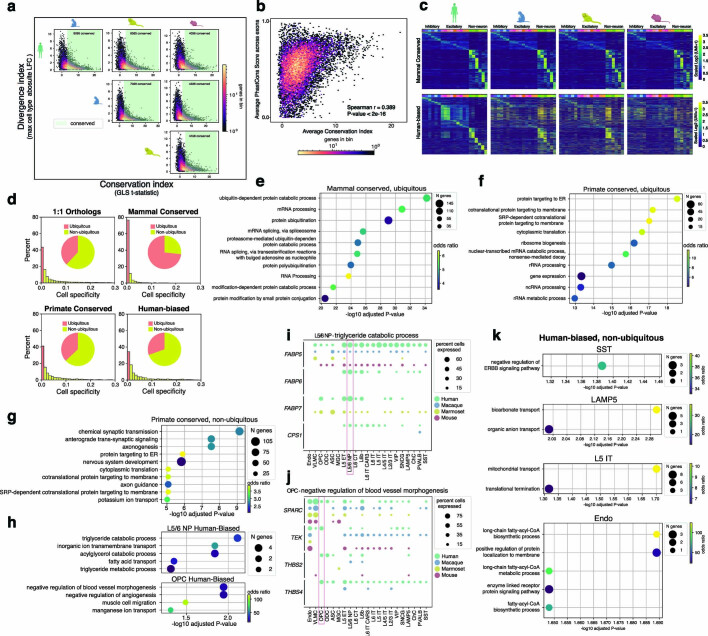
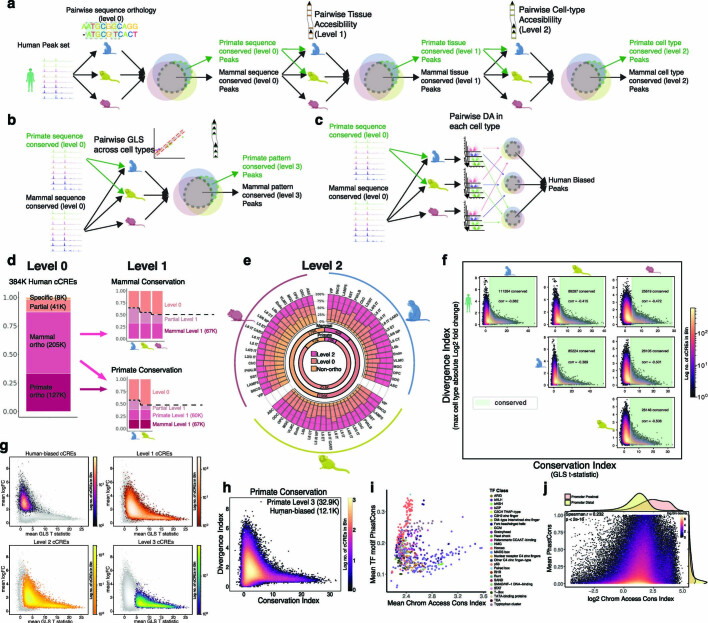
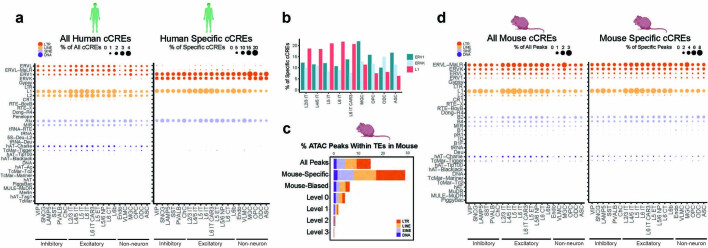
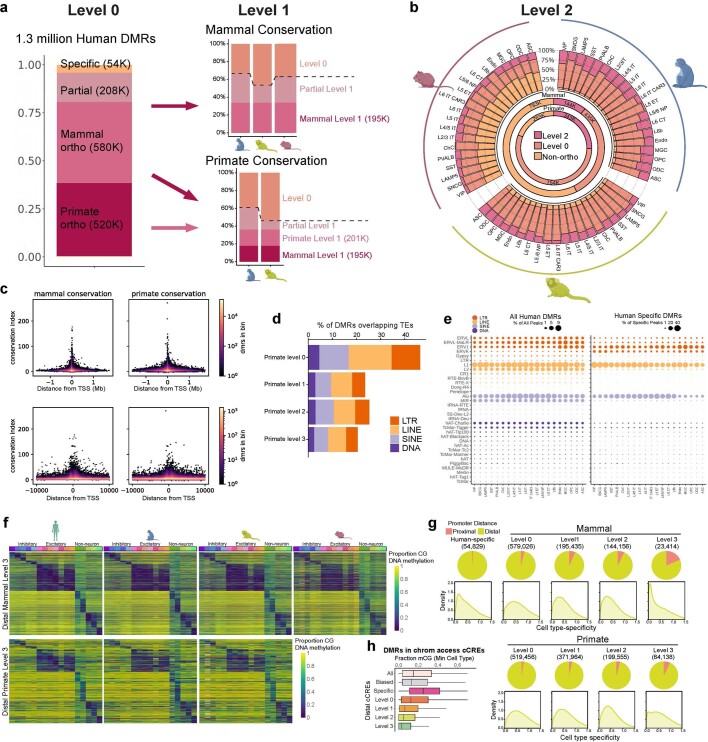
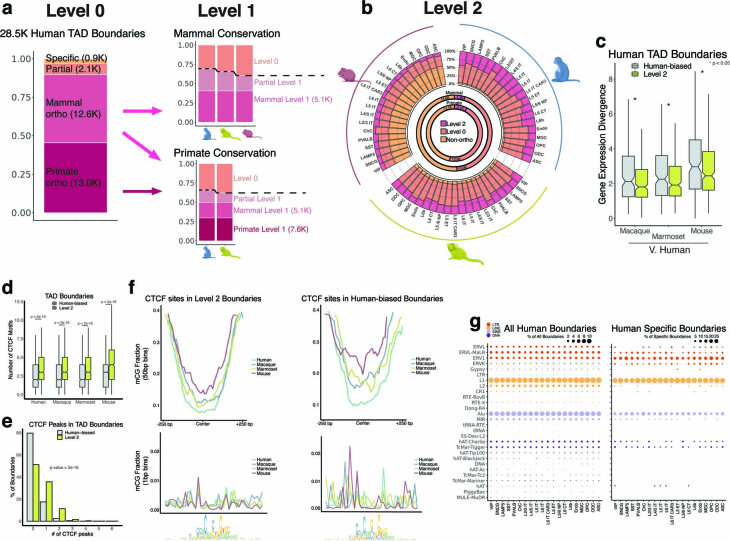
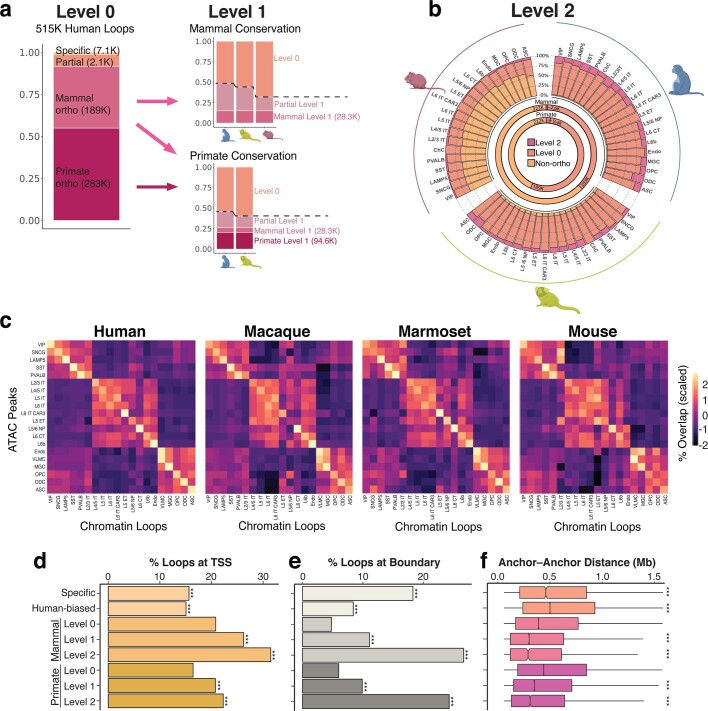
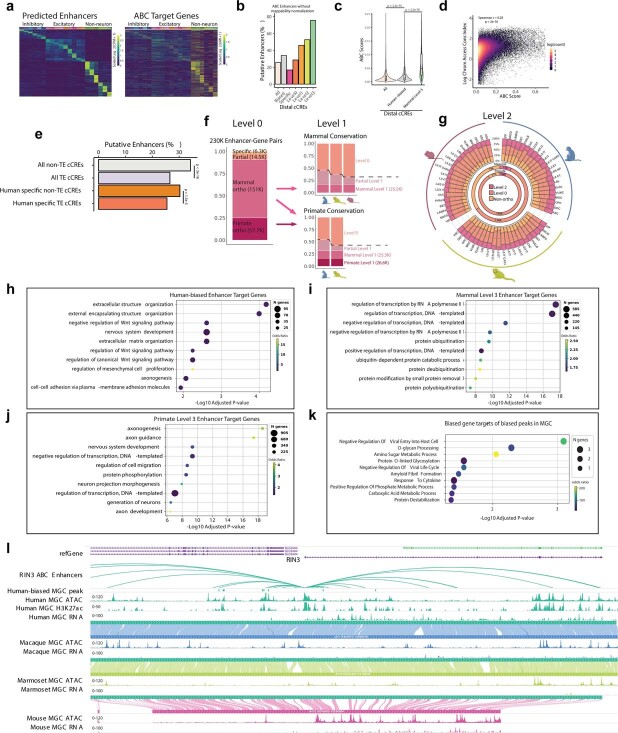
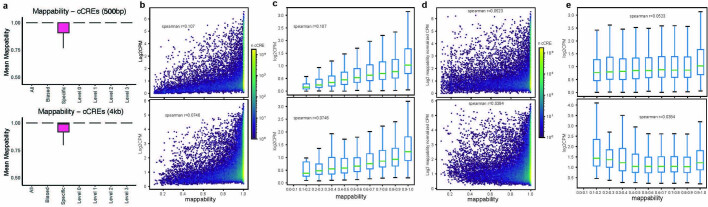
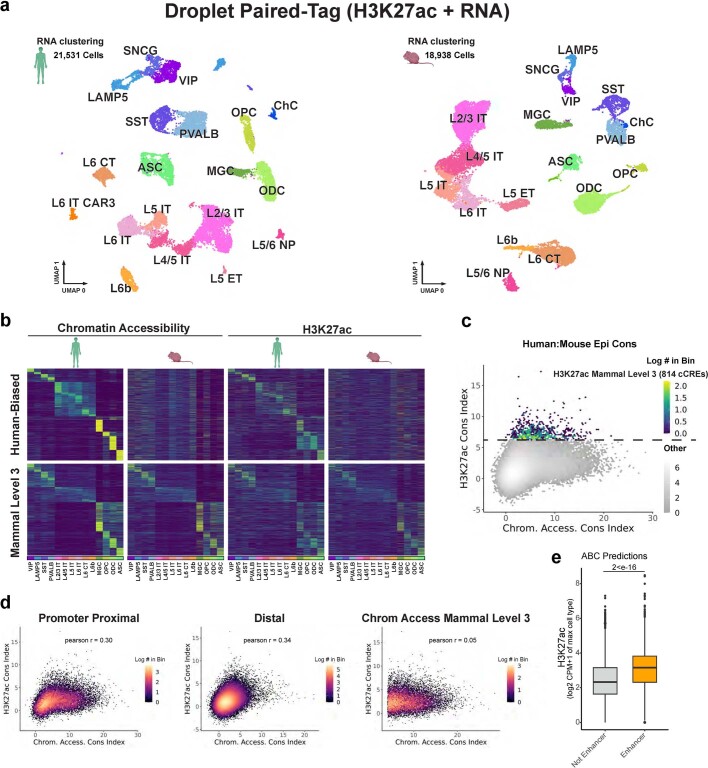
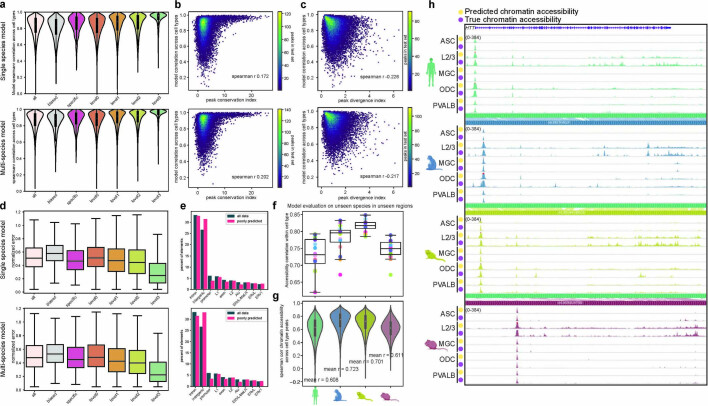
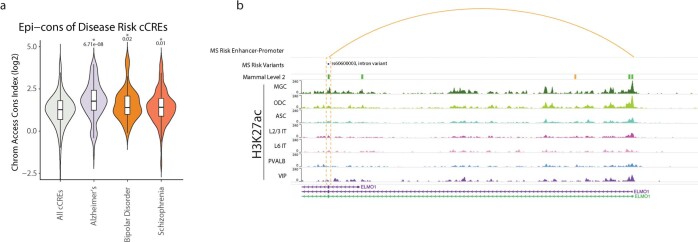
Divergence of cis-regulatory elements drives species-specific traits1, but how this manifests in the evolution of the neocortex at the molecular and cellular level remains unclear. Here we investigated the gene regulatory programs in the primary motor cortex of human, macaque, marmoset and mouse using single-cell multiomics assays, generating gene expression, chromatin accessibility, DNA methylome and chromosomal conformation profiles from a total of over 200,000 cells. From these data, we show evidence that divergence of transcription factor expression corresponds to species-specific epigenome landscapes. We find that conserved and divergent gene regulatory features are reflected in the evolution of the three-dimensional genome. Transposable elements contribute to nearly 80% of the human-specific candidate cis-regulatory elements in cortical cells. Through machine learning, we develop sequence-based predictors of candidate cis-regulatory elements in different species and demonstrate that the genomic regulatory syntax is highly preserved from rodents to primates. Finally, we show that epigenetic conservation combined with sequence similarity helps to uncover functional cis-regulatory elements and enhances our ability to interpret genetic variants contributing to neurological disease and traits.
© 2023. The Author(s).
Conflict of interest statement
B.R. is a co-founder and consultant of Arima Genomics and co-founder of Epigenome Technologies. J.R.E. is on the scientific advisory board of Zymo Research.
Figures
References
-
- Scanning Human Gene Deserts for Long-range Enhancers (Lawrence Berkeley National Laboratory, 2003). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
