

Brain-wide correspondence of neuronal epigenomics and distant projections
- PMID: 38092919
- PMCID: PMC10719087
- DOI: 10.1038/s41586-023-06823-w
Brain-wide correspondence of neuronal epigenomics and distant projections
Abstract
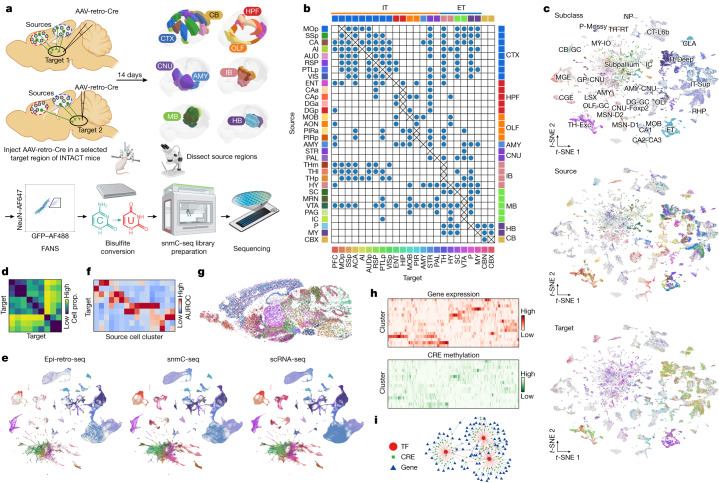
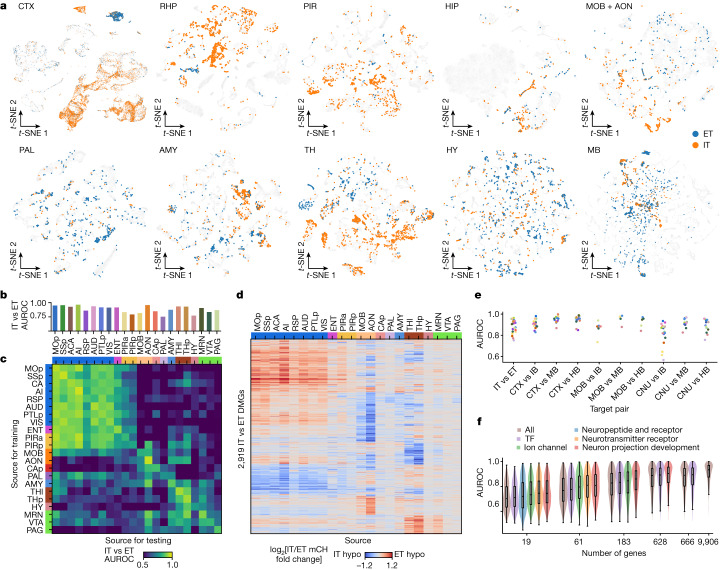
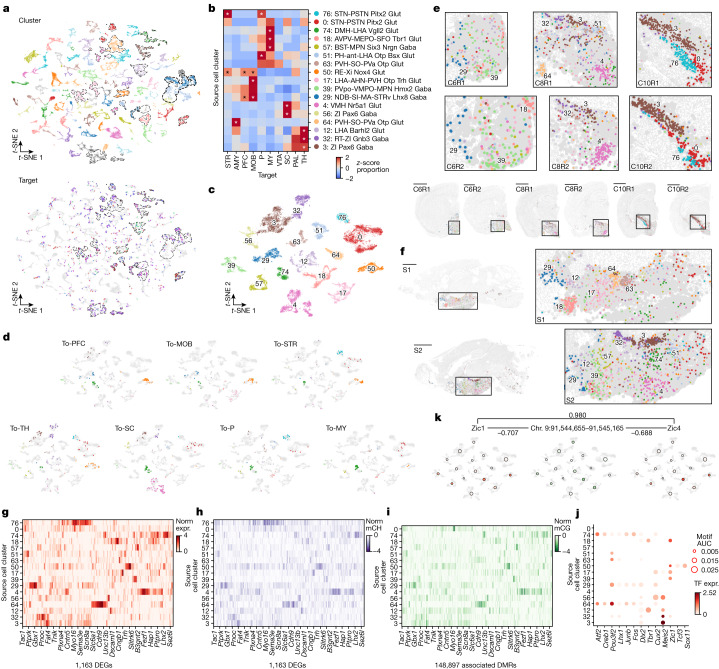
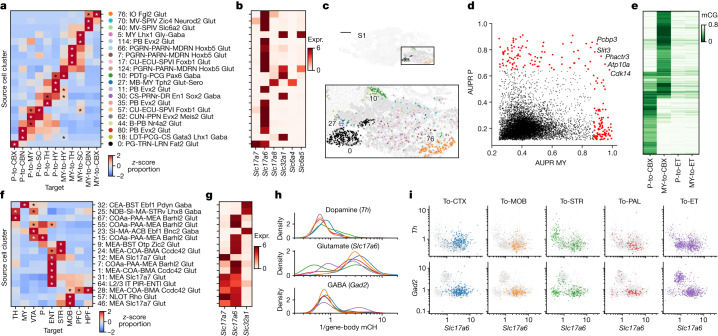
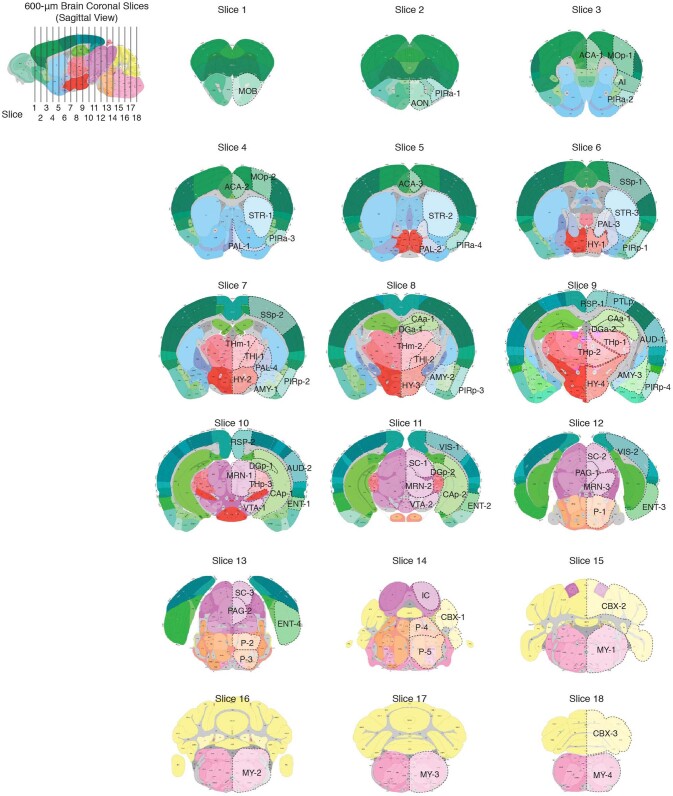
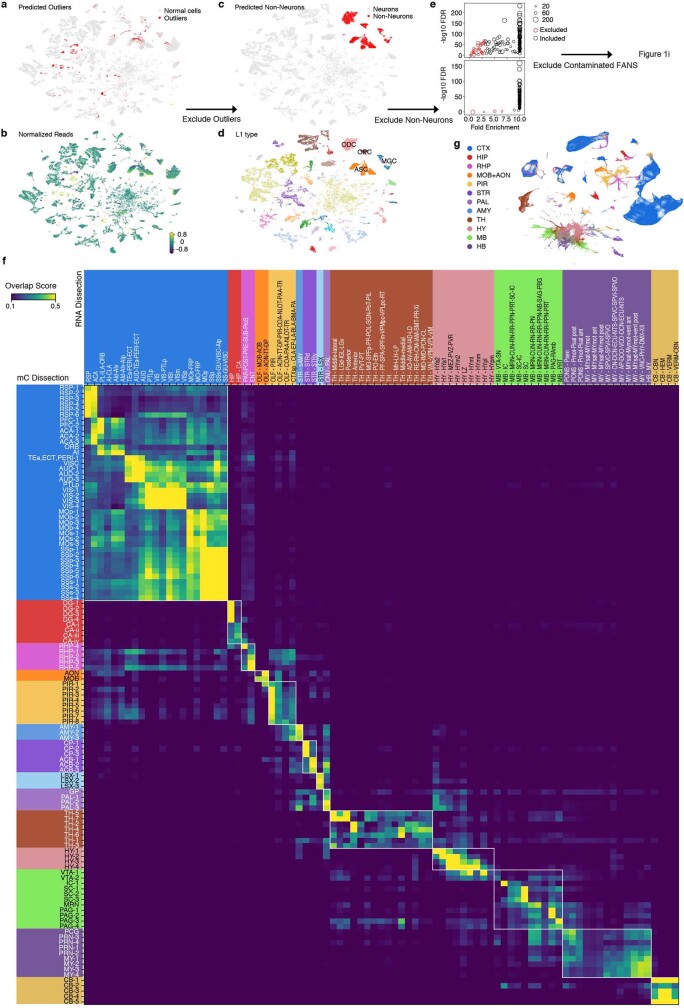
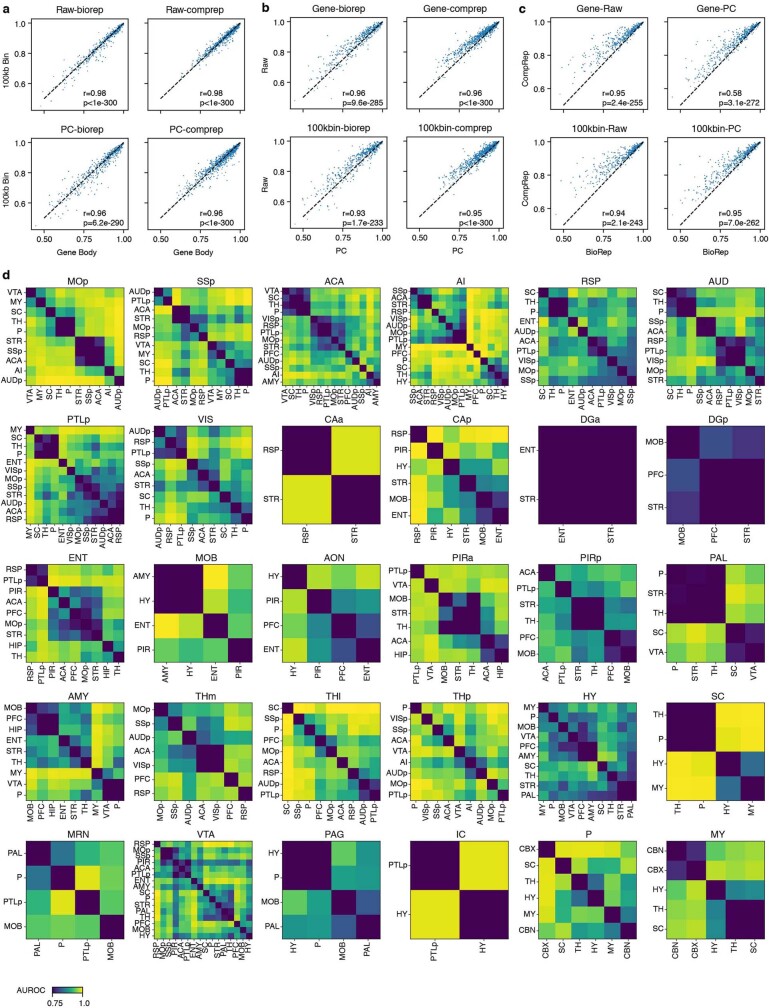
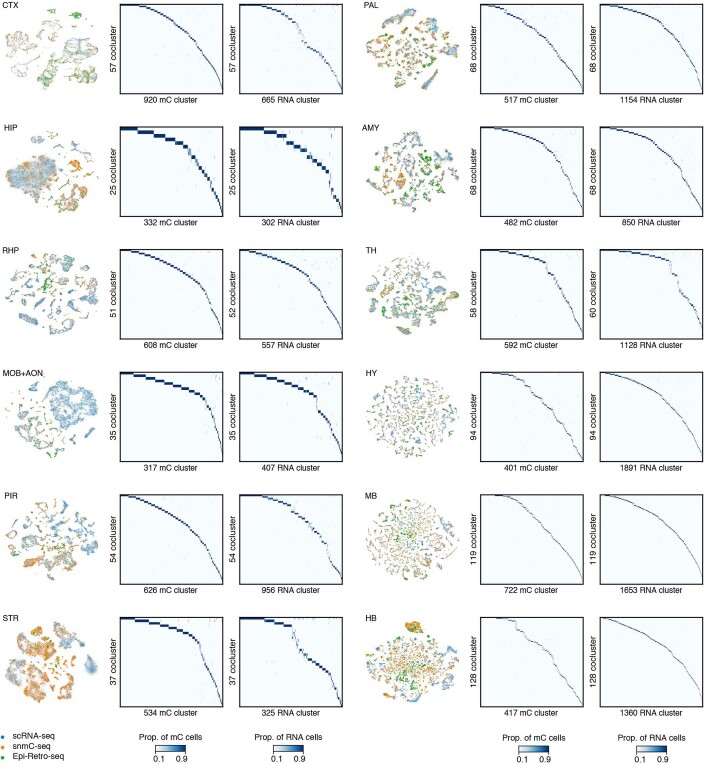
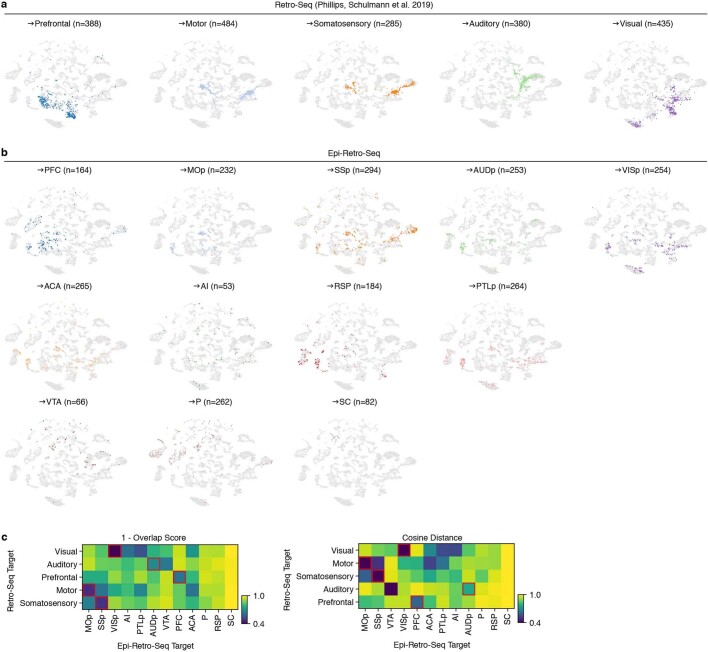
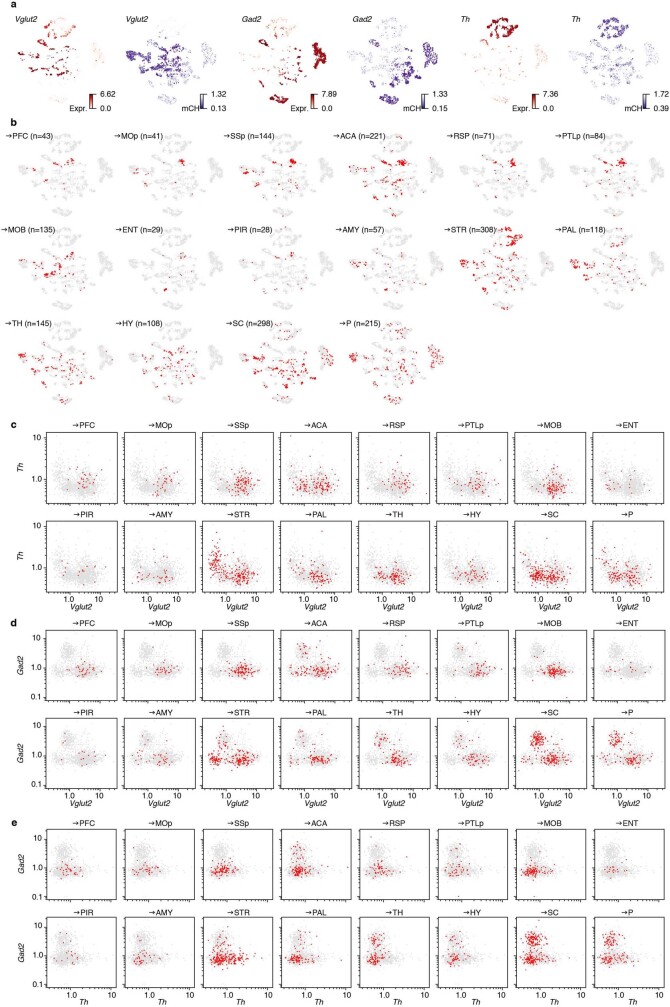
Single-cell analyses parse the brain's billions of neurons into thousands of 'cell-type' clusters residing in different brain structures1. Many cell types mediate their functions through targeted long-distance projections allowing interactions between specific cell types. Here we used epi-retro-seq2 to link single-cell epigenomes and cell types to long-distance projections for 33,034 neurons dissected from 32 different regions projecting to 24 different targets (225 source-to-target combinations) across the whole mouse brain. We highlight uses of these data for interrogating principles relating projection types to transcriptomics and epigenomics, and for addressing hypotheses about cell types and connections related to genetics. We provide an overall synthesis with 926 statistical comparisons of discriminability of neurons projecting to each target for every source. We integrate this dataset into the larger BRAIN Initiative Cell Census Network atlas, composed of millions of neurons, to link projection cell types to consensus clusters. Integration with spatial transcriptomics further assigns projection-enriched clusters to smaller source regions than the original dissections. We exemplify this by presenting in-depth analyses of projection neurons from the hypothalamus, thalamus, hindbrain, amygdala and midbrain to provide insights into properties of those cell types, including differentially expressed genes, their associated cis-regulatory elements and transcription-factor-binding motifs, and neurotransmitter use.
© 2023. The Author(s).
Conflict of interest statement
J.R.E. serves on the scientific advisory board of Zymo Research Inc.
Figures


Comment in
-
Cellular atlases of the entire mouse brain.Nature. 2023 Dec;624(7991):253-255. doi: 10.1038/d41586-023-03781-1. Nature. 2023. PMID: 38092903 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
