

Slide-tags enables single-nucleus barcoding for multimodal spatial genomics
- PMID: 38093010
- PMCID: PMC10764288
- DOI: 10.1038/s41586-023-06837-4
Slide-tags enables single-nucleus barcoding for multimodal spatial genomics
Erratum in
-
Publisher Correction: Slide-tags enables single-nucleus barcoding for multimodal spatial genomics.Nature. 2024 Jan;625(7994):E11. doi: 10.1038/s41586-023-06961-1. Nature. 2024. PMID: 38110579 Free PMC article. No abstract available.
Abstract
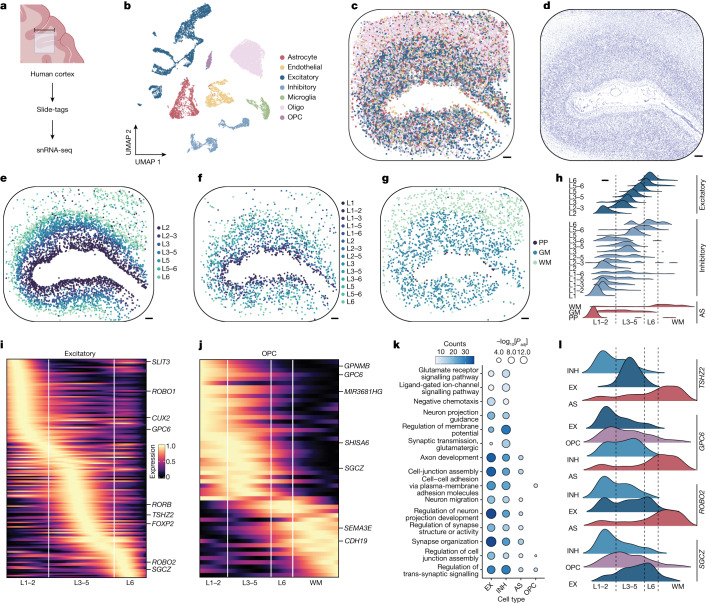
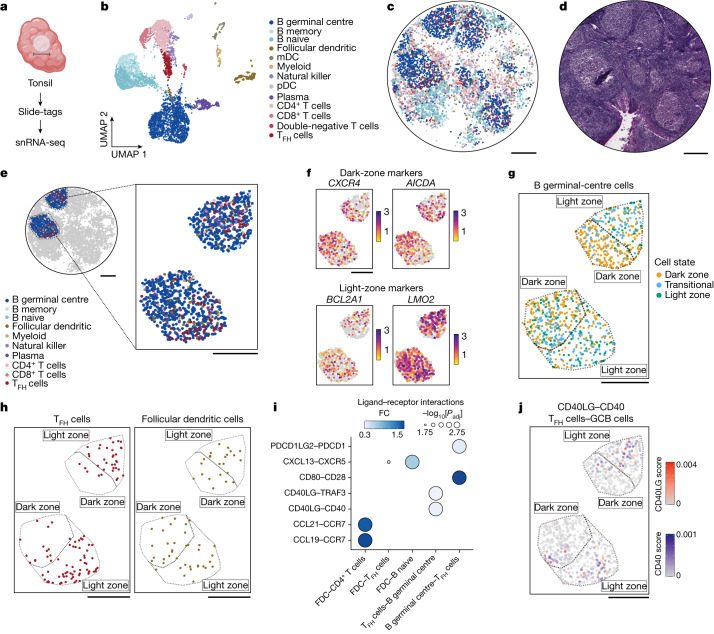
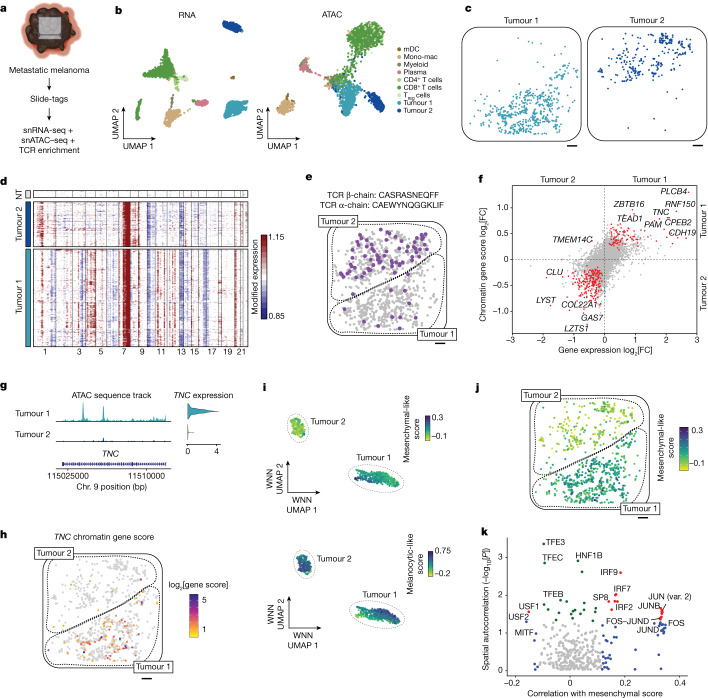
Recent technological innovations have enabled the high-throughput quantification of gene expression and epigenetic regulation within individual cells, transforming our understanding of how complex tissues are constructed1-6. However, missing from these measurements is the ability to routinely and easily spatially localize these profiled cells. We developed a strategy, Slide-tags, in which single nuclei within an intact tissue section are tagged with spatial barcode oligonucleotides derived from DNA-barcoded beads with known positions. These tagged nuclei can then be used as an input into a wide variety of single-nucleus profiling assays. Application of Slide-tags to the mouse hippocampus positioned nuclei at less than 10 μm spatial resolution and delivered whole-transcriptome data that are indistinguishable in quality from ordinary single-nucleus RNA-sequencing data. To demonstrate that Slide-tags can be applied to a wide variety of human tissues, we performed the assay on brain, tonsil and melanoma. We revealed cell-type-specific spatially varying gene expression across cortical layers and spatially contextualized receptor-ligand interactions driving B cell maturation in lymphoid tissue. A major benefit of Slide-tags is that it is easily adaptable to almost any single-cell measurement technology. As a proof of principle, we performed multiomic measurements of open chromatin, RNA and T cell receptor (TCR) sequences in the same cells from metastatic melanoma, identifying transcription factor motifs driving cancer cell state transitions in spatially distinct microenvironments. Slide-tags offers a universal platform for importing the compendium of established single-cell measurements into the spatial genomics repertoire.
© 2023. The Author(s).
Conflict of interest statement
E.Z.M. and F.C. are academic founders of Curio Bioscience. F.C. is an academic co-founder of Doppler Bio and an advisor to Amber Bio. F.C., E.Z.M., A.J.C.R., J.A.W., N.M.N. and V.K. are listed as inventors on a patent application related to the work. C.J.W. holds equity in BioNTech. The other authors declare no competing interests.
Figures











Update of
-
Slide-tags: scalable, single-nucleus barcoding for multi-modal spatial genomics.bioRxiv [Preprint]. 2023 Apr 3:2023.04.01.535228. doi: 10.1101/2023.04.01.535228. bioRxiv. 2023. Update in: Nature. 2024 Jan;625(7993):101-109. doi: 10.1038/s41586-023-06837-4. PMID: 37066158 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
