

Genetic Spectrum of Polycystic Kidney and Liver Diseases and the Resulting Phenotypes
- PMID: 38097330
- PMCID: PMC10746289
- DOI: 10.1053/j.akdh.2023.04.004
Genetic Spectrum of Polycystic Kidney and Liver Diseases and the Resulting Phenotypes
Abstract
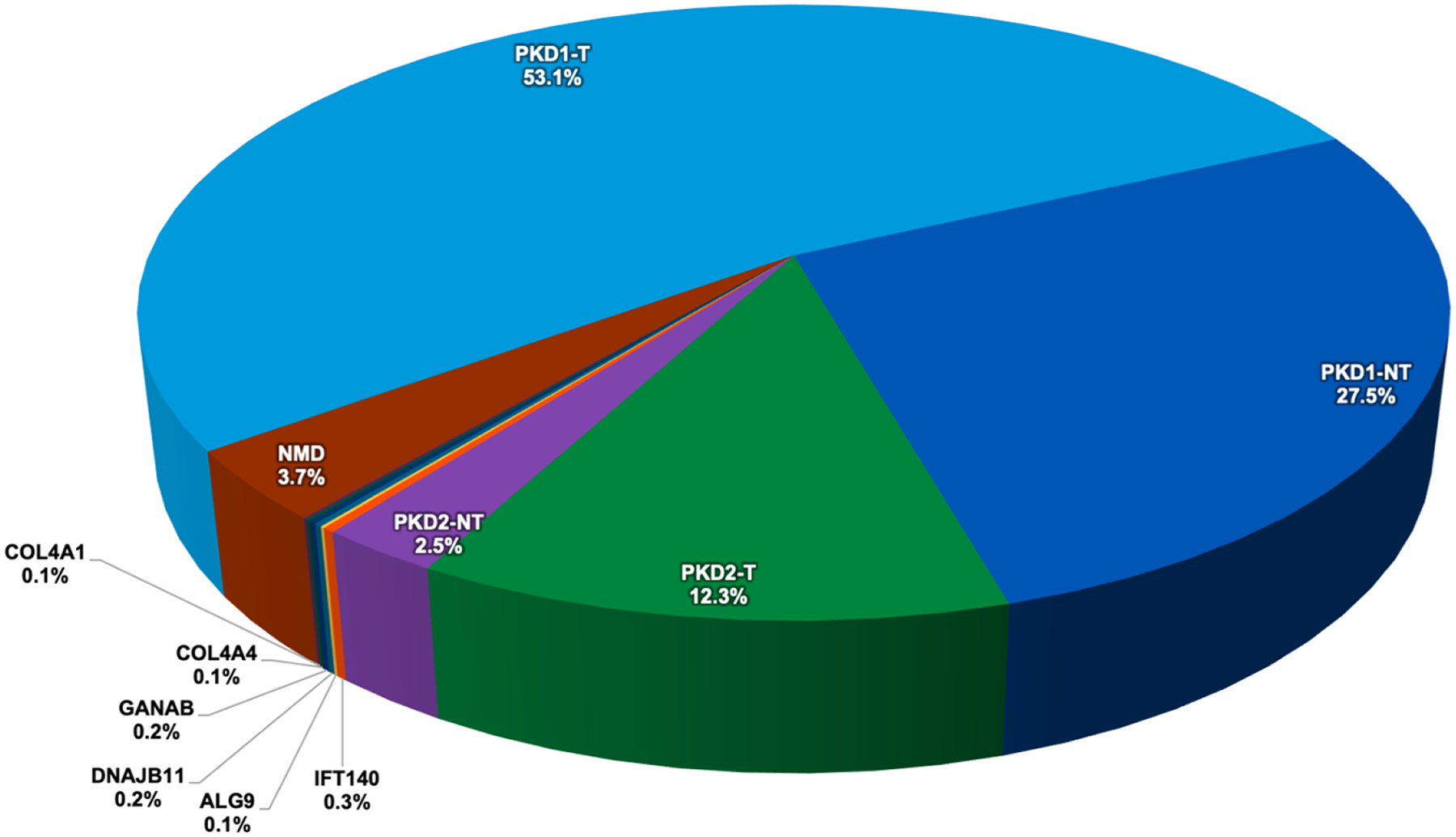
Polycystic kidney diseases are a group of monogenically inherited disorders characterized by cyst development in the kidney with defects in primary cilia function central to pathogenesis. Autosomal dominant polycystic kidney disease (ADPKD) has progressive cystogenesis and accounts for 5-10% of kidney failure (KF) patients. There are two major ADPKD genes, PKD1 and PKD2, and seven minor loci. PKD1 accounts for ∼80% of patients and is associated with the most severe disease (KF is typically at 55-65 years); PKD2 accounts for ∼15% of families, with KF typically in the mid-70s. The minor genes are generally associated with milder kidney disease, but for DNAJB11 and ALG5, the age at KF is similar to PKD2. PKD1 and PKD2 have a high level of allelic heterogeneity, with no single pathogenic variant accounting for >2% of patients. Additional genetic complexity includes biallelic disease, sometimes causing very early-onset ADPKD, and mosaicism. Autosomal dominant polycystic liver disease is characterized by severe PLD but limited PKD. The two major genes are PRKCSH and SEC63, while GANAB, ALG8, and PKHD1 can present as ADPKD or autosomal dominant polycystic liver disease. Autosomal recessive polycystic kidney disease typically has an infantile onset, with PKHD1 being the major locus and DZIP1L and CYS1 being minor genes. In addition, there are a range of mainly recessive syndromic ciliopathies with PKD as part of the phenotype. Because of the phenotypic and genic overlap between the diseases, employing a next-generation sequencing panel containing all known PKD and ciliopathy genes is recommended for clinical testing.
Keywords: ADPKD; ADPLD; ARPKD; Genetics; Polycystic kidney disease.
Copyright © 2023 National Kidney Foundation, Inc. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
P. Harris reports the following:
Consultancy: Vertex, Mitobridge, Regulus, Otsuka, Janssen, Maze Therapeutics, CorrectorBio
Figures


References
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
