

Beyond the exome: utility of long-read whole genome sequencing in exome-negative autosomal recessive diseases
- PMID: 38098057
- PMCID: PMC10720148
- DOI: 10.1186/s13073-023-01270-8
Beyond the exome: utility of long-read whole genome sequencing in exome-negative autosomal recessive diseases
Abstract
Background: Long-read whole genome sequencing (lrWGS) has the potential to address the technical limitations of exome sequencing in ways not possible by short-read WGS. However, its utility in autosomal recessive Mendelian diseases is largely unknown.
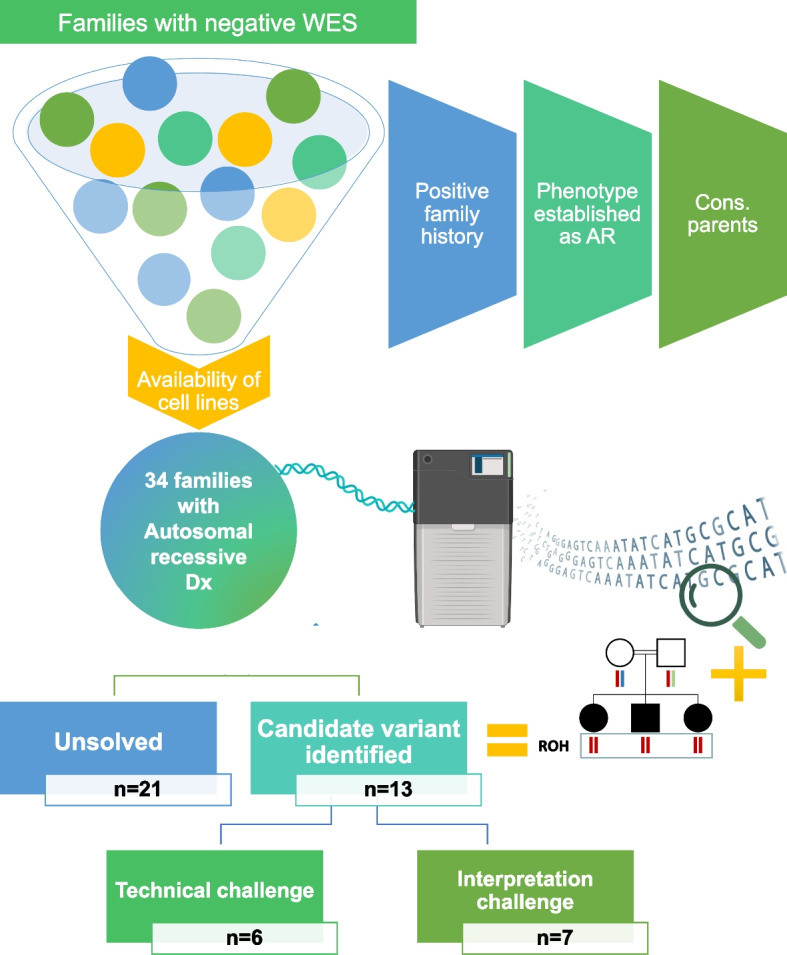
Methods: In a cohort of 34 families in which the suspected autosomal recessive diseases remained undiagnosed by exome sequencing, lrWGS was performed on the Pacific Bioscience Sequel IIe platform.
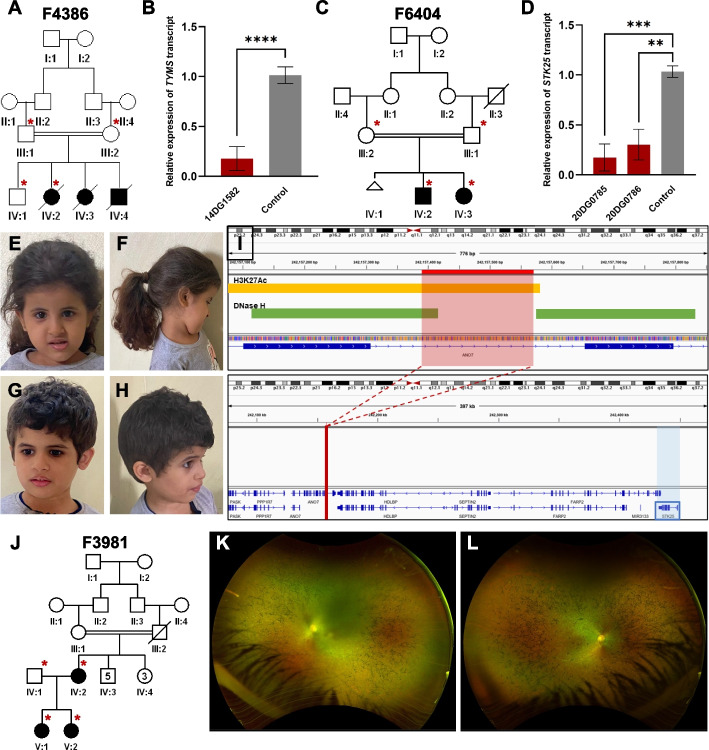
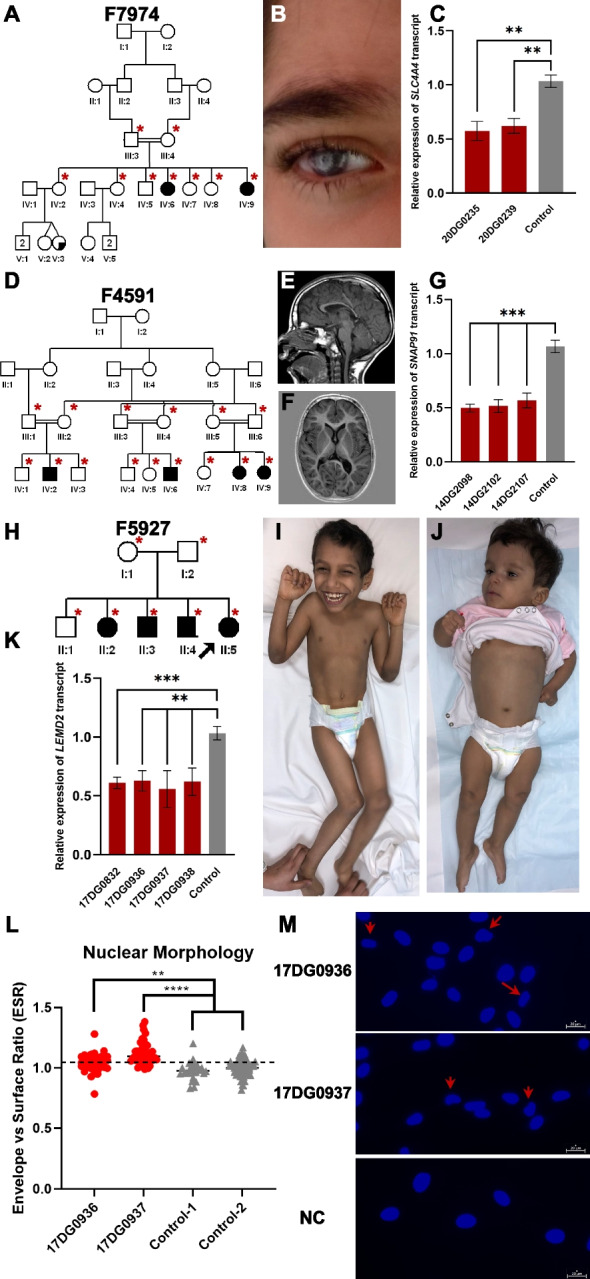
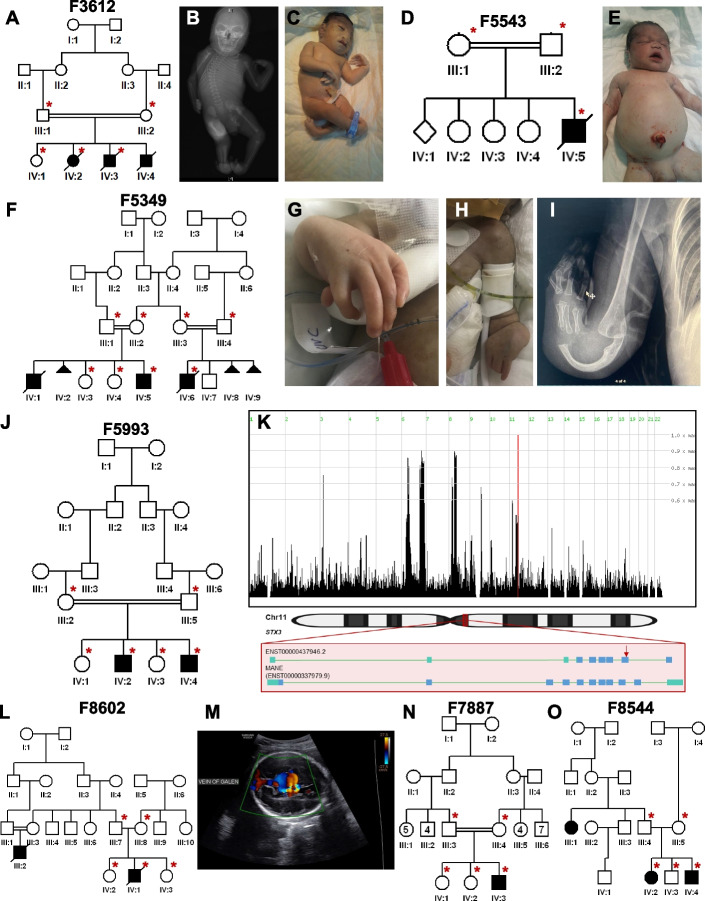
Results: Likely causal variants were identified in 13 (38%) of the cohort. These include (1) a homozygous splicing SV in TYMS as a novel candidate gene for lethal neonatal lactic acidosis, (2) a homozygous non-coding SV that we propose impacts STK25 expression and causes a novel neurodevelopmental disorder, (3) a compound heterozygous SV in RP1L1 with complex inheritance pattern in a family with inherited retinal disease, (4) homozygous deep intronic variants in LEMD2 and SNAP91 as novel candidate genes for neurodevelopmental disorders in two families, and (5) a promoter SNV in SLC4A4 causing non-syndromic band keratopathy. Surprisingly, we also encountered causal variants that could have been identified by short-read exome sequencing in 7 families. The latter highlight scenarios that are especially challenging at the interpretation level.
Conclusions: Our data highlight the continued need to address the interpretation challenges in parallel with efforts to improve the sequencing technology itself. We propose a path forward for the implementation of lrWGS sequencing in the setting of autosomal recessive diseases in a way that maximizes its utility.
Keywords: ABHD12; Autozygome; C1orf109; FLVCR1; Long-read sequencing; NID1; PKHD1; SHFM; STX3.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Alkuraya FS. How the human genome transformed study of rare diseases. Nature. 2021;590:218–9. - PubMed
-
- Lowther C, Valkanas E, Giordano JL, Wang HZ, Currall BB, O'Keefe K, et al. Systematic evaluation of genome sequencing for the diagnostic assessment of autism spectrum disorder and fetal structural anomalies. Am J Hum Genet. 2023;110(9):1454–1469. doi: 10.1016/j.ajhg.2023.07.010. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
