

A time-resolved meta-analysis of consensus gene expression profiles during human T-cell activation
- PMID: 38098113
- PMCID: PMC10722659
- DOI: 10.1186/s13059-023-03120-7
A time-resolved meta-analysis of consensus gene expression profiles during human T-cell activation
Abstract
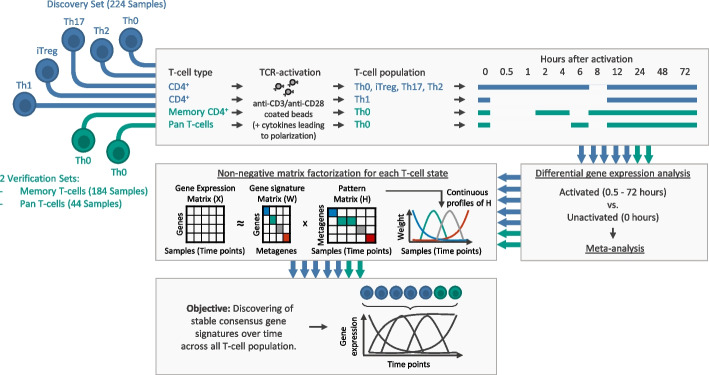
Background: The coordinated transcriptional regulation of activated T-cells is based on a complex dynamic behavior of signaling networks. Given an external stimulus, T-cell gene expression is characterized by impulse and sustained patterns over the course. Here, we analyze the temporal pattern of activation across different T-cell populations to develop consensus gene signatures for T-cell activation.
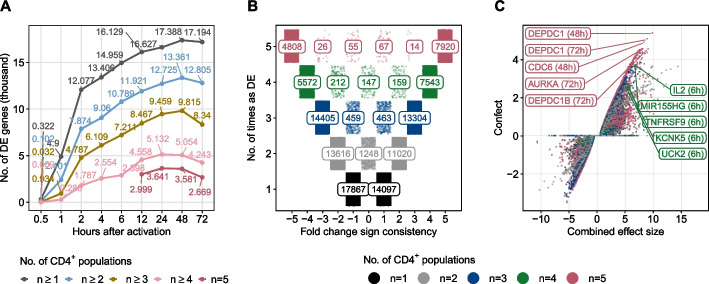
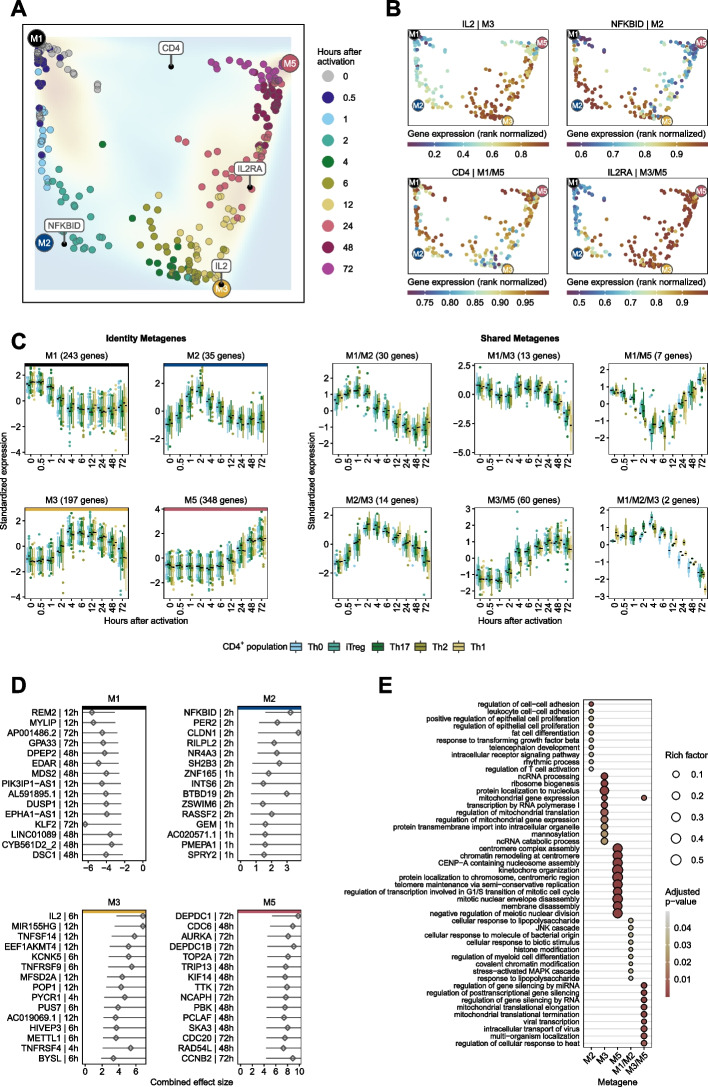
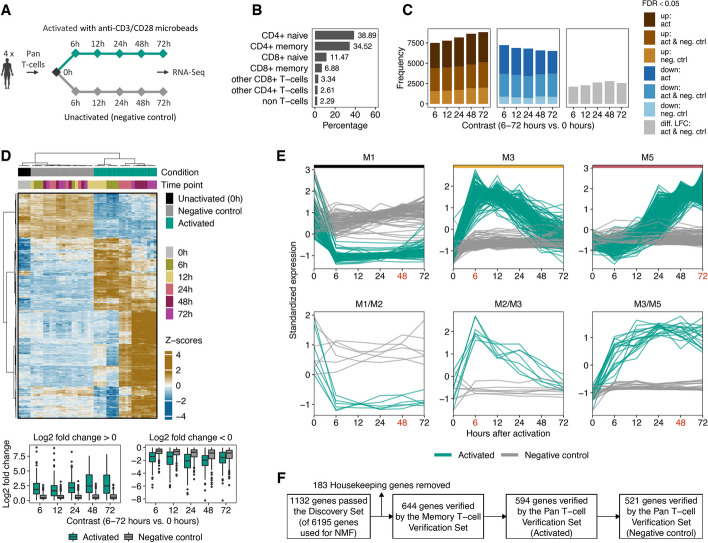
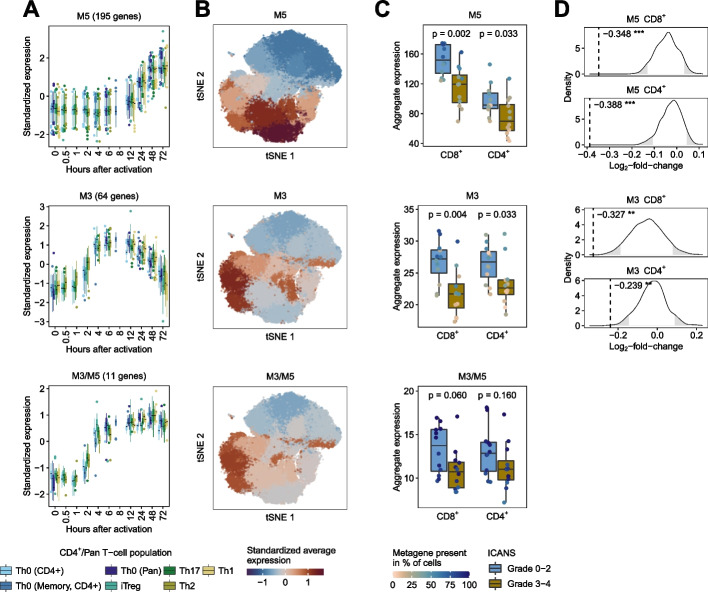
Results: Here, we identify and verify general biomarker signatures robustly evaluating T-cell activation in a time-resolved manner. We identify time-resolved gene expression profiles comprising 521 genes of up to 10 disjunct time points during activation and different polarization conditions. The gene signatures include central transcriptional regulators of T-cell activation, representing successive waves as well as sustained patterns of induction. They cover sustained repressed, intermediate, and late response expression rates across multiple T-cell populations, thus defining consensus biomarker signatures for T-cell activation. In addition, intermediate and late response activation signatures in CAR T-cell infusion products are correlated to immune effector cell-associated neurotoxicity syndrome.
Conclusion: This study is the first to describe temporally resolved gene expression patterns across T-cell populations. These biomarker signatures are a valuable source, e.g., monitoring transcriptional changes during T-cell activation with a reasonable number of genes, annotating T-cell states in single-cell transcriptome studies, or assessing dysregulated functions of human T-cell immunity.
Keywords: Biomarkers; Gene expression; Non-negative matrix factorization; T-cell activation; Temporal gene profiles; Time series; Transcriptome.
© 2023. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
