

Rational design of a SOCS1-edited tumor-infiltrating lymphocyte therapy using CRISPR/Cas9 screens
- PMID: 38099496
- PMCID: PMC10721144
- DOI: 10.1172/JCI163096
Rational design of a SOCS1-edited tumor-infiltrating lymphocyte therapy using CRISPR/Cas9 screens
Abstract
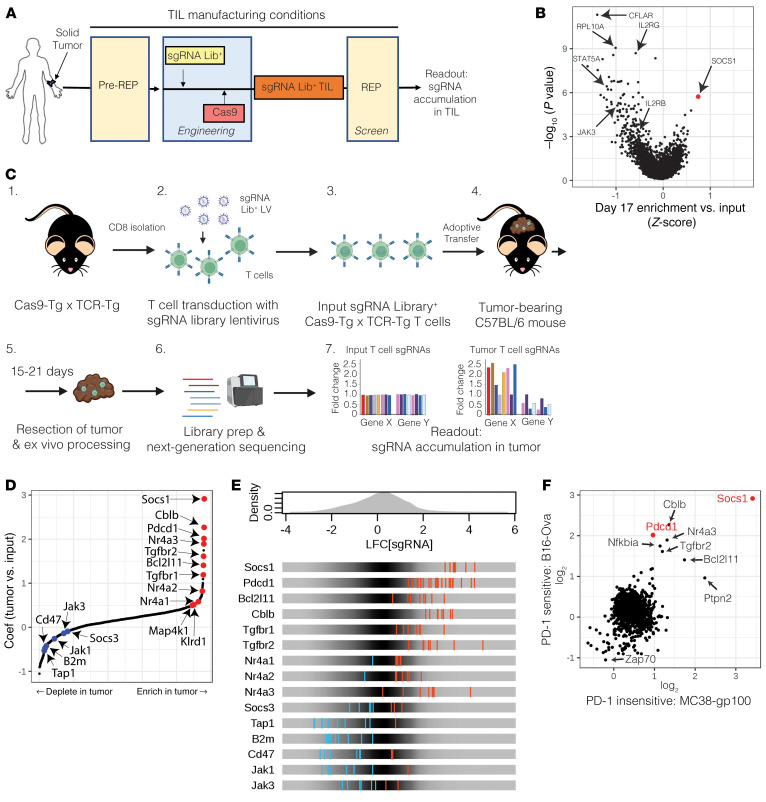
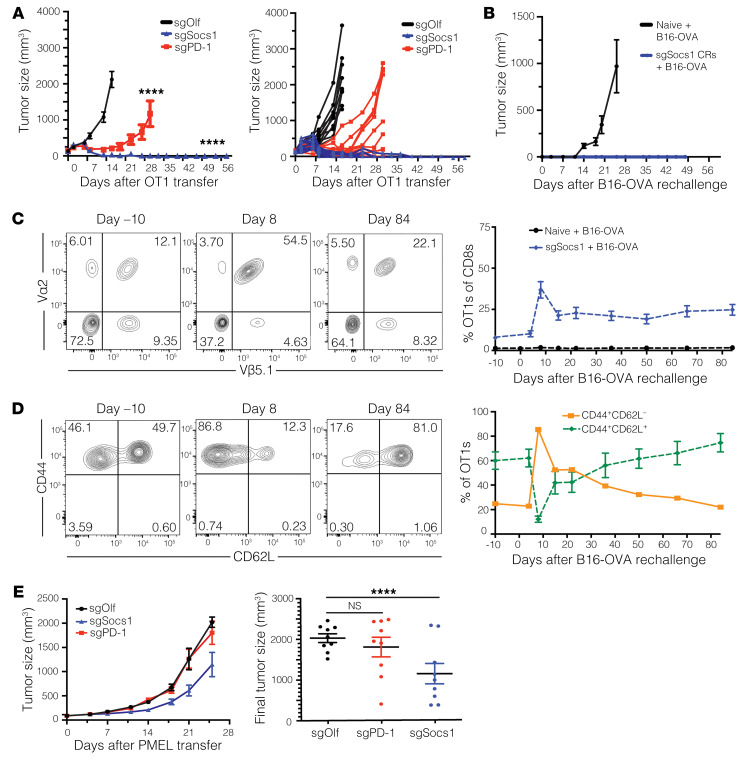
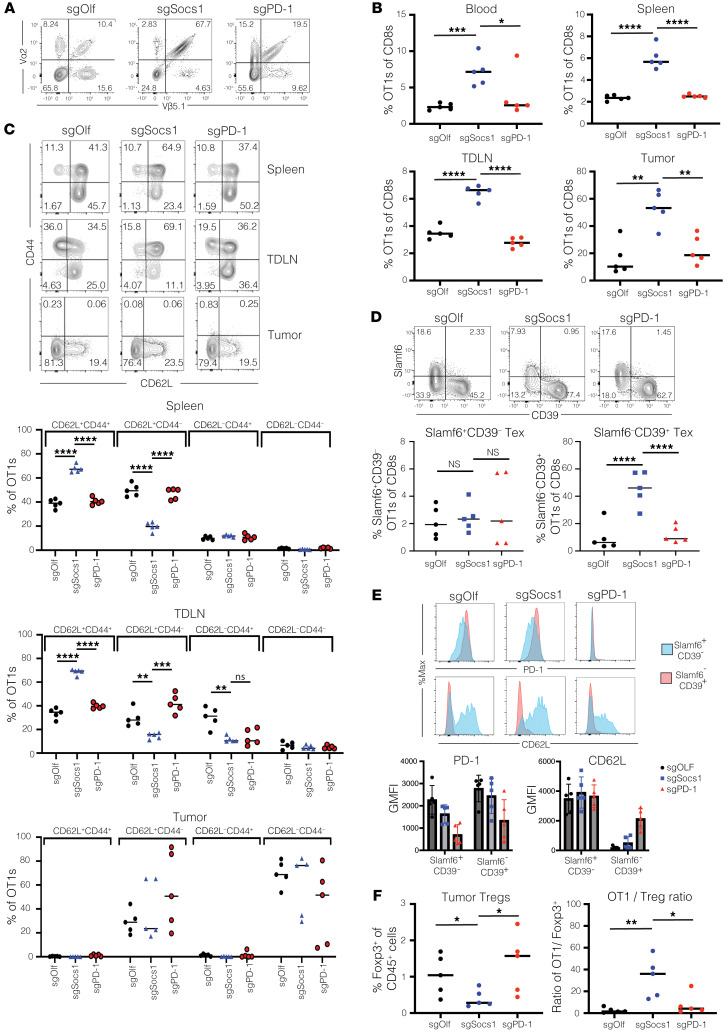
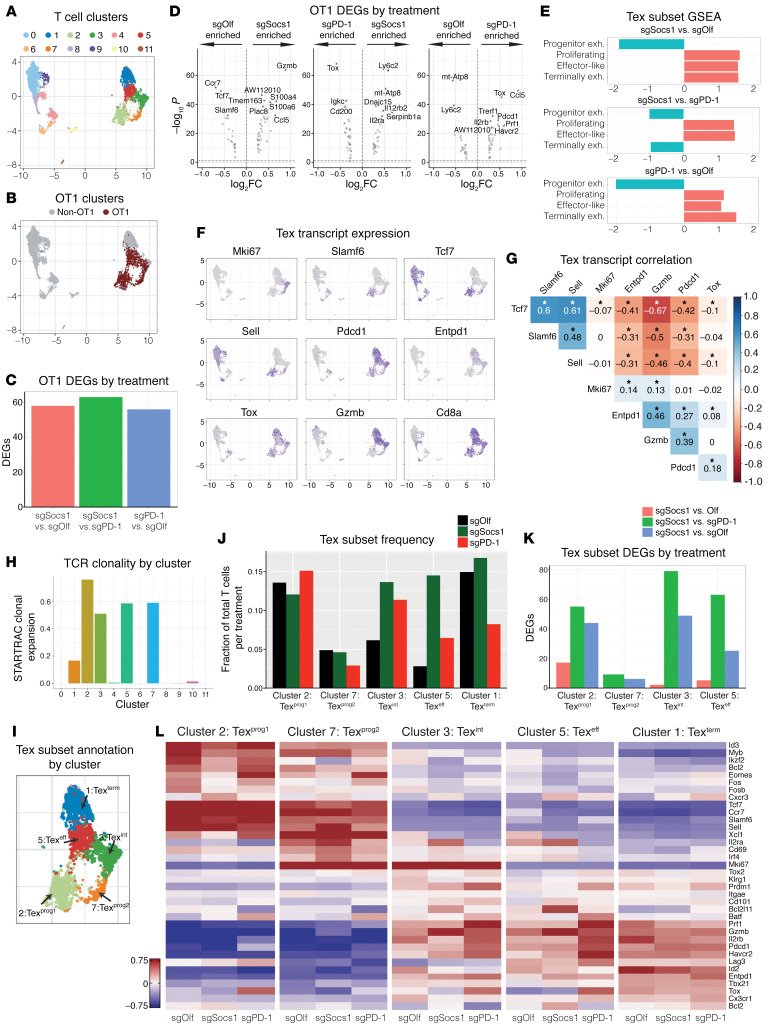
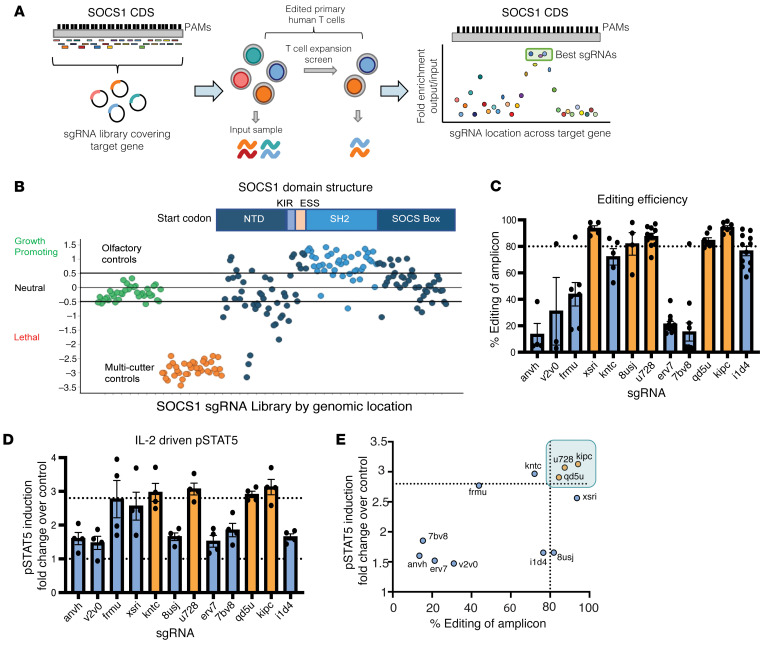
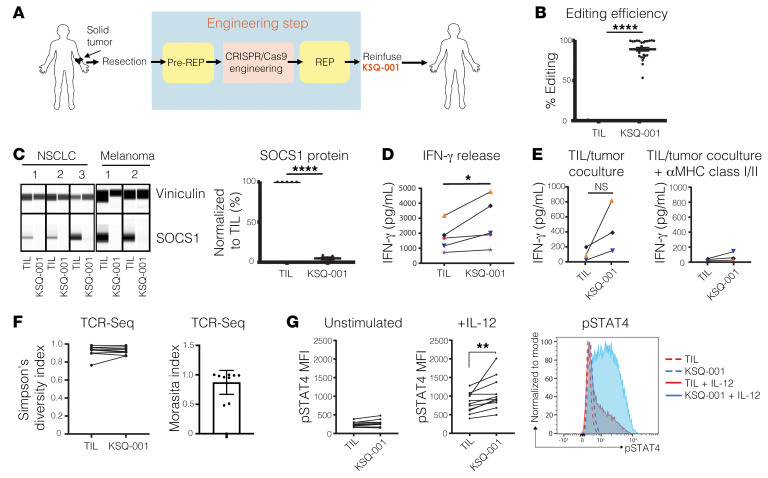
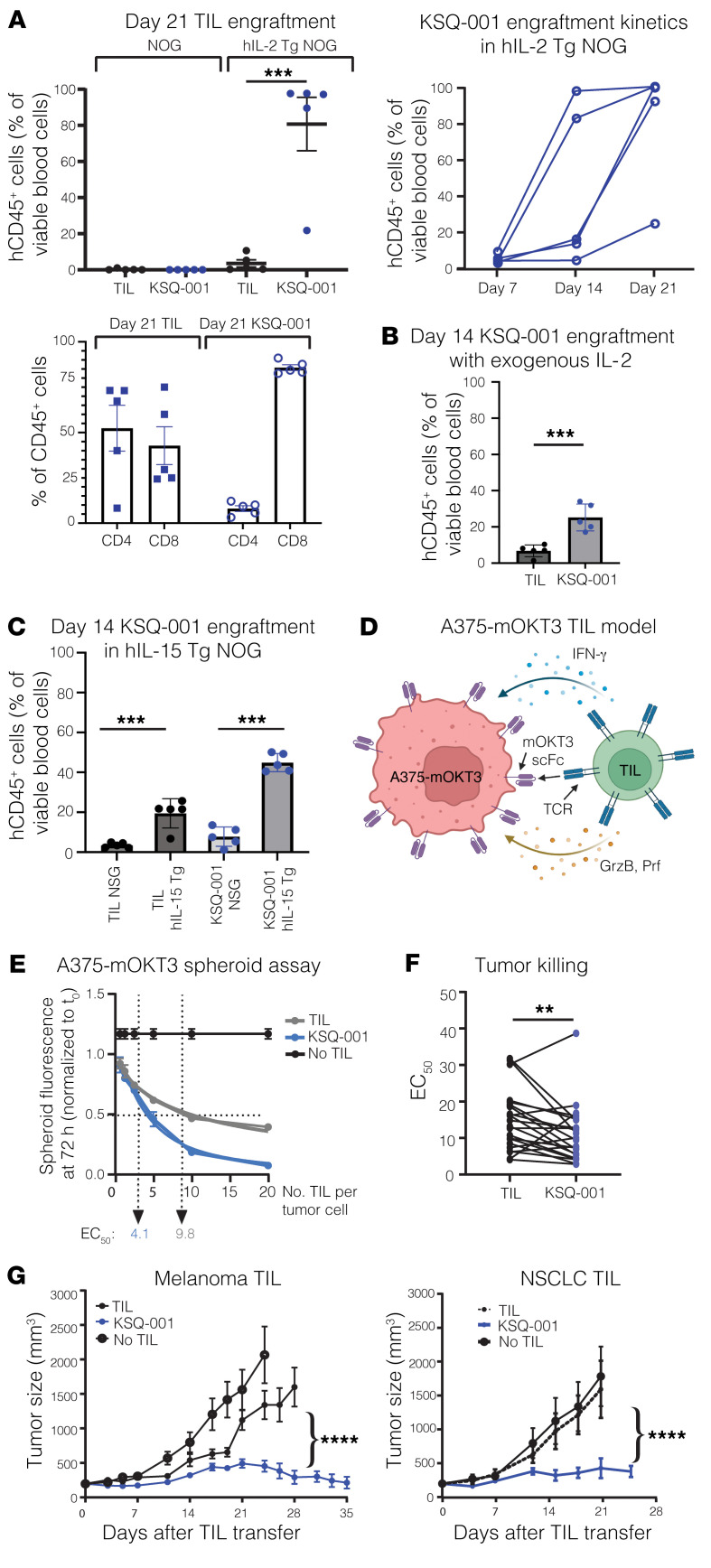
Cell therapies such as tumor-infiltrating lymphocyte (TIL) therapy have shown promise in the treatment of patients with refractory solid tumors, with improvement in response rates and durability of responses nevertheless sought. To identify targets capable of enhancing the antitumor activity of T cell therapies, large-scale in vitro and in vivo clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 screens were performed, with the SOCS1 gene identified as a top T cell-enhancing target. In murine CD8+ T cell-therapy models, SOCS1 served as a critical checkpoint in restraining the accumulation of central memory T cells in lymphoid organs as well as intermediate (Texint) and effector (Texeff) exhausted T cell subsets derived from progenitor exhausted T cells (Texprog) in tumors. A comprehensive CRISPR tiling screen of the SOCS1-coding region identified sgRNAs targeting the SH2 domain of SOCS1 as the most potent, with an sgRNA with minimal off-target cut sites used to manufacture KSQ-001, an engineered TIL therapy with SOCS1 inactivated by CRISPR/Cas9. KSQ-001 possessed increased responsiveness to cytokine signals and enhanced in vivo antitumor function in mouse models. These data demonstrate the use of CRISPR/Cas9 screens in the rational design of T cell therapies.
Keywords: Cancer immunotherapy; Gene therapy; Immunology; T cells; Therapeutics.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
