

Empagliflozin treatment rescues abnormally reduced Na+ currents in ventricular cardiomyocytes from dystrophin-deficient mdx mice
- PMID: 38099845
- PMCID: PMC11219046
- DOI: 10.1152/ajpheart.00729.2023
Empagliflozin treatment rescues abnormally reduced Na+ currents in ventricular cardiomyocytes from dystrophin-deficient mdx mice
Abstract
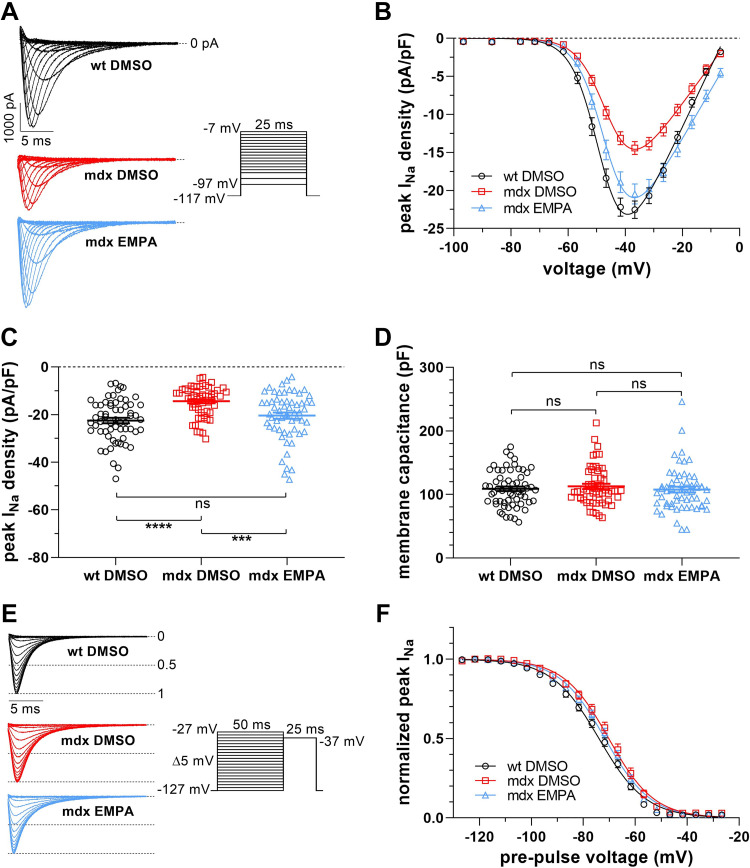
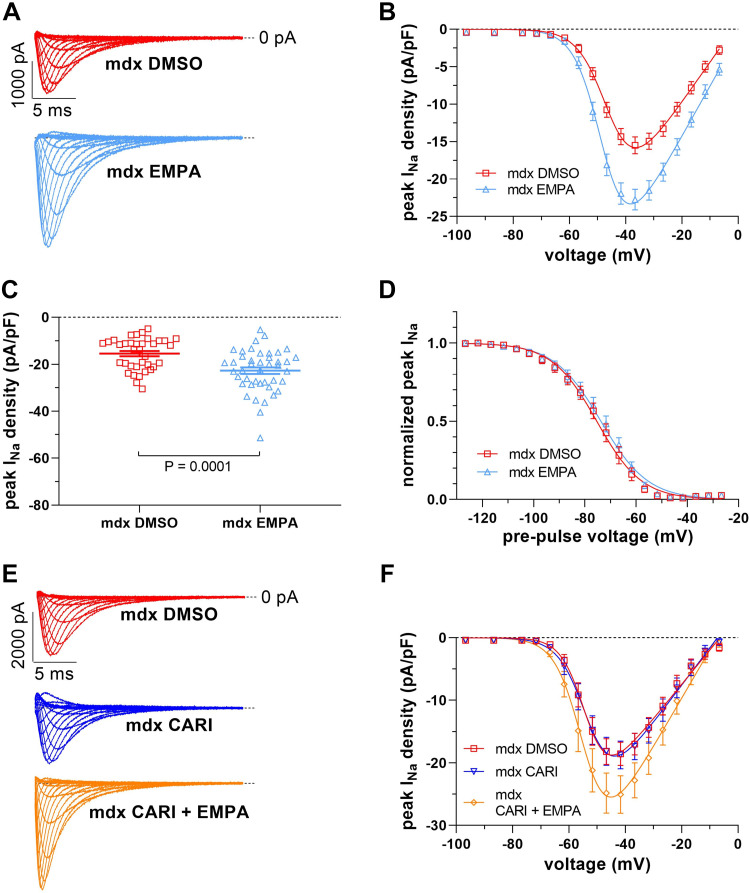
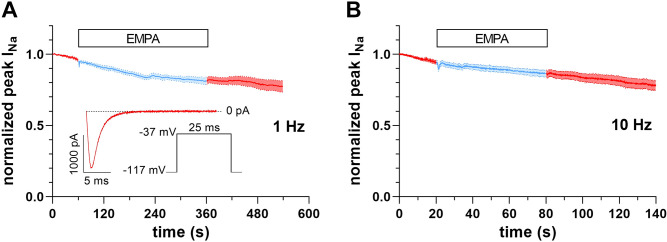
Cardiac arrhythmias significantly contribute to mortality in Duchenne muscular dystrophy (DMD), a severe muscle illness caused by mutations in the gene encoding for the intracellular protein dystrophin. A major source for arrhythmia vulnerability in patients with DMD is impaired ventricular impulse conduction, which predisposes for ventricular asynchrony, decreased cardiac output, and the development of reentrant circuits. Using the dystrophin-deficient mdx mouse model for human DMD, we previously reported that the lack of dystrophin causes a significant loss of peak Na+ current (INa) in ventricular cardiomyocytes. This finding provided a mechanistic explanation for ventricular conduction defects and concomitant arrhythmias in the dystrophic heart. In the present study, we explored the hypothesis that empagliflozin (EMPA), an inhibitor of sodium/glucose cotransporter 2 in clinical use to treat type II diabetes and nondiabetic heart failure, rescues peak INa loss in dystrophin-deficient ventricular cardiomyocytes. We found that INa of cardiomyocytes derived from mdx mice, which had received clinically relevant doses of EMPA for 4 wk, was restored to wild-type level. Moreover, incubation of isolated mdx ventricular cardiomyocytes with 1 µM EMPA for 24 h significantly increased their peak INa. This effect was independent of Na+-H+ exchanger 1 inhibition by the drug. Our findings imply that EMPA treatment can rescue abnormally reduced peak INa of dystrophin-deficient ventricular cardiomyocytes. Long-term EMPA administration may diminish arrhythmia vulnerability in patients with DMD.NEW & NOTEWORTHY Dystrophin deficiency in cardiomyocytes leads to abnormally reduced Na+ currents. These can be rescued by long-term empagliflozin treatment.
Keywords: Duchenne muscular dystrophy; arrhythmias; cardiomyocyte sodium currents; empagliflozin; mdx mice.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical