

Heterogeneity and transcriptional drivers of triple-negative breast cancer
- PMID: 38100350
- PMCID: PMC10842760
- DOI: 10.1016/j.celrep.2023.113564
Heterogeneity and transcriptional drivers of triple-negative breast cancer
Abstract
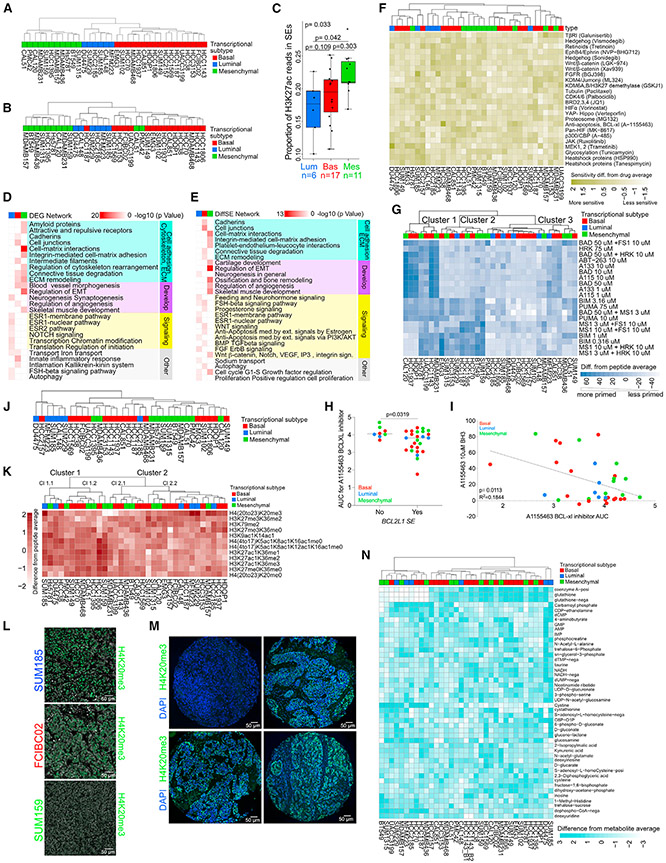
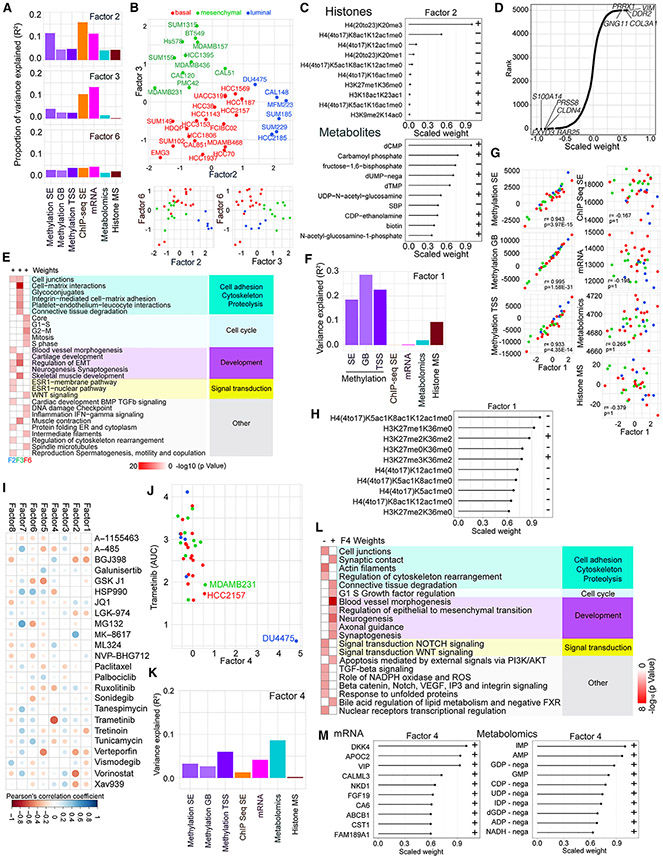
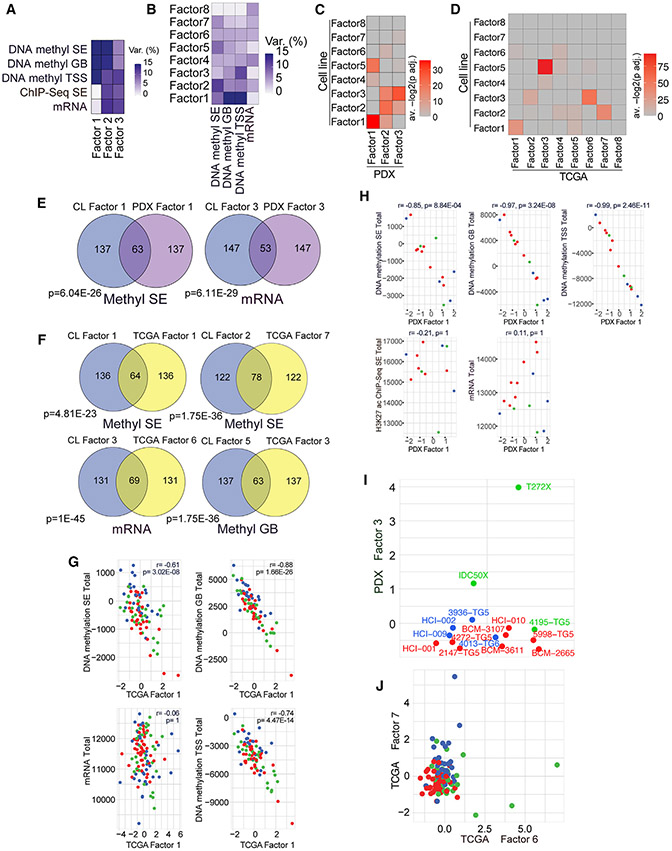
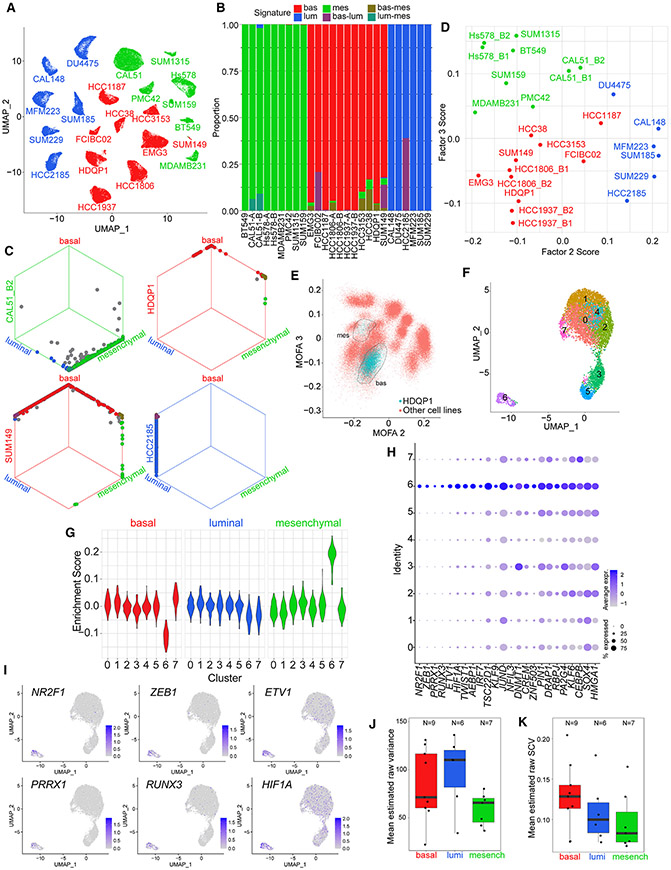
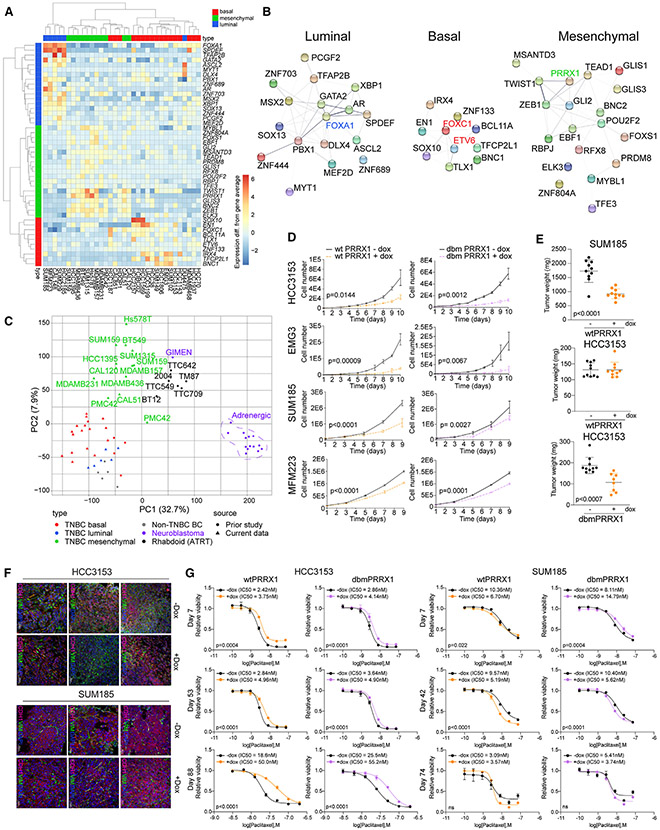
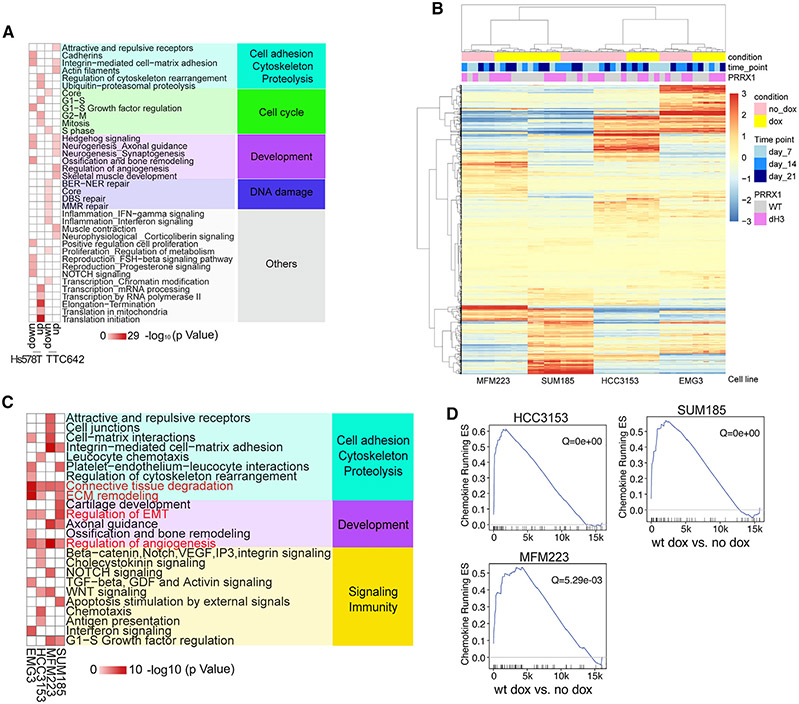
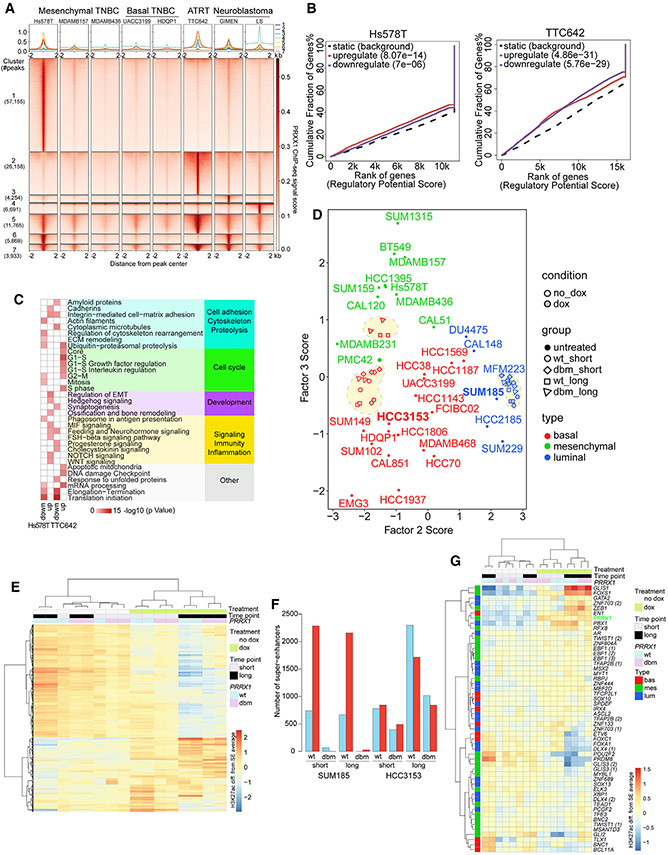
Triple-negative breast cancer (TNBC) is a heterogeneous disease with limited treatment options. To characterize TNBC heterogeneity, we defined transcriptional, epigenetic, and metabolic subtypes and subtype-driving super-enhancers and transcription factors by combining functional and molecular profiling with computational analyses. Single-cell RNA sequencing revealed relative homogeneity of the major transcriptional subtypes (luminal, basal, and mesenchymal) within samples. We found that mesenchymal TNBCs share features with mesenchymal neuroblastoma and rhabdoid tumors and that the PRRX1 transcription factor is a key driver of these tumors. PRRX1 is sufficient for inducing mesenchymal features in basal but not in luminal TNBC cells via reprogramming super-enhancer landscapes, but it is not required for mesenchymal state maintenance or for cellular viability. Our comprehensive, large-scale, multiplatform, multiomics study of both experimental and clinical TNBC is an important resource for the scientific and clinical research communities and opens venues for future investigation.
Keywords: CP: Cancer; triple-negative breast cancer; tumor heterogeneity.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The following authors report current employment: Eli Lilly (B.J.), Shasqi, Inc (M.A.), GenieUsGenomics (A.T.), Morrison & Foerster LLP (A.G.), AstraZeneca (M.B.E. and L.E.S.), Odyssey Therapeutics (J.D.J.). K.P. serves on the Scientific Advisory Boards (SABs) of Novartis, Ideaya Biosciences, and Scorpion Therapeutics; holds equity options in Scorpion Therapeutics and Ideaya Biosciences; and receives sponsored research funding from Novartis, where she consults. F.M. is a cofounder of and has equity in Harbinger Health, has equity in Zephyr AI, and consults for Harbinger Health and Zephyr AI. She is on the board of directors of Exscientia Plc. She declares that none of these relationships are directly or indirectly related to the content of this manuscript. P.S. is a consultant for Novartis, Genovis, Guidepoint, The Planning Shop, ORIC Pharmaceuticals, Cedilla Therapeutics, Syros Pharmaceuticals, Blueprint Medicines, Curie Bio, Differentiated Therapeutics, Excientia, Ligature Therapeutics, Merck, Redesign Science, Sibylla Biotech, and Exo Therapeutics; he receives research funding from Novartis. A.G.L. serves on the SAB of Flash Therapeutics, Zentalis Pharmaceuticals, and Trueline Therapeutics and consults for AbbVie. M.B. receives research funding from Novartis, where he also serves on the SAB and acts as a consultant. He is a member of the SAB for Kronos Bio and GV20 Therapeutics and holds equity in both companies. He also serves on the SAB for FibroGen and is a consultant for Belharra Therapeutics. K.W.W. serves on the SAB of TScan Therapeutics, SQZ Biotech, Bisou Bioscience Company, DEM BioPharma, and Nextechinvest; receives sponsored research funding from Novartis; and is a co-founder, stockholder, and advisory board member of Immunitas Therapeutics. D.D. receives research support from Canon, Inc. H.W.L. receives research funding from Novartis.
Figures
Similar articles
-
Profiling triple-negative breast cancer-specific super-enhancers identifies high-risk mesenchymal development subtype and BETi-Targetable vulnerabilities.Mol Cancer. 2025 May 13;24(1):141. doi: 10.1186/s12943-025-02342-6. Mol Cancer. 2025. PMID: 40361105 Free PMC article.
-
RNA binding protein RBMS3 is a common EMT effector that modulates triple-negative breast cancer progression via stabilizing PRRX1 mRNA.Oncogene. 2021 Nov;40(46):6430-6442. doi: 10.1038/s41388-021-02030-x. Epub 2021 Oct 4. Oncogene. 2021. PMID: 34608266 Free PMC article.
-
Expression of epithelial-mesenchymal transition-related markers in triple-negative breast cancer: ZEB1 as a potential biomarker for poor clinical outcome.Hum Pathol. 2015 Sep;46(9):1267-74. doi: 10.1016/j.humpath.2015.05.010. Epub 2015 May 30. Hum Pathol. 2015. PMID: 26170011
-
Long non-coding RNAs: implications in targeted diagnoses, prognosis, and improved therapeutic strategies in human non- and triple-negative breast cancer.Clin Epigenetics. 2018 Jun 27;10:88. doi: 10.1186/s13148-018-0514-z. eCollection 2018. Clin Epigenetics. 2018. PMID: 29983835 Free PMC article. Review.
-
Triple-negative breast cancer and basal-like subtype : Pathology and targeted therapy.J Med Invest. 2021;68(3.4):213-219. doi: 10.2152/jmi.68.213. J Med Invest. 2021. PMID: 34759133 Review.
Cited by
-
BET inhibitors (BETi) influence oxidative phosphorylation metabolism by affecting mitochondrial dynamics leading to alterations in apoptotic pathways in triple-negative breast cancer (TNBC) cells.Cell Prolif. 2024 Dec;57(12):e13730. doi: 10.1111/cpr.13730. Epub 2024 Sep 2. Cell Prolif. 2024. PMID: 39223828 Free PMC article.
-
FOSL1 transcriptionally dictates the Warburg effect and enhances chemoresistance in triple-negative breast cancer.J Transl Med. 2025 Jan 2;23(1):1. doi: 10.1186/s12967-024-06014-9. J Transl Med. 2025. PMID: 39748430 Free PMC article.
-
Comparative analysis of statistical and deep learning-based multi-omics integration for breast cancer subtype classification.J Transl Med. 2025 Jul 1;23(1):709. doi: 10.1186/s12967-025-06662-5. J Transl Med. 2025. PMID: 40598554 Free PMC article.
-
Metabolic shifts in lipid utilization and reciprocal interactions within the lung metastatic niche of triple-negative breast cancer revealed by spatial multi-omics.Cell Death Dis. 2024 Dec 18;15(12):899. doi: 10.1038/s41419-024-07205-4. Cell Death Dis. 2024. PMID: 39695088 Free PMC article.
-
CREB5 promotes tumorigenicity and upregulates druggable cell surface modalities in basal-like breast cancer.NPJ Precis Oncol. 2025 Aug 6;9(1):276. doi: 10.1038/s41698-025-01051-z. NPJ Precis Oncol. 2025. PMID: 40770411 Free PMC article.
References
-
- Su Y, Subedee A, Bloushtain-Qimron N, Savova V, Krzystanek M, Li L, Marusyk A, Tabassum DP, Zak A, Flacker MJ, et al. (2015). Somatic Cell Fusions Reveal Extensive Heterogeneity in Basal-like Breast Cancer. Cell Rep. 11, 1549–1563. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
