

Whole-genome sequencing reveals the molecular implications of the stepwise progression of lung adenocarcinoma
- PMID: 38102134
- PMCID: PMC10724178
- DOI: 10.1038/s41467-023-43732-y
Whole-genome sequencing reveals the molecular implications of the stepwise progression of lung adenocarcinoma
Abstract
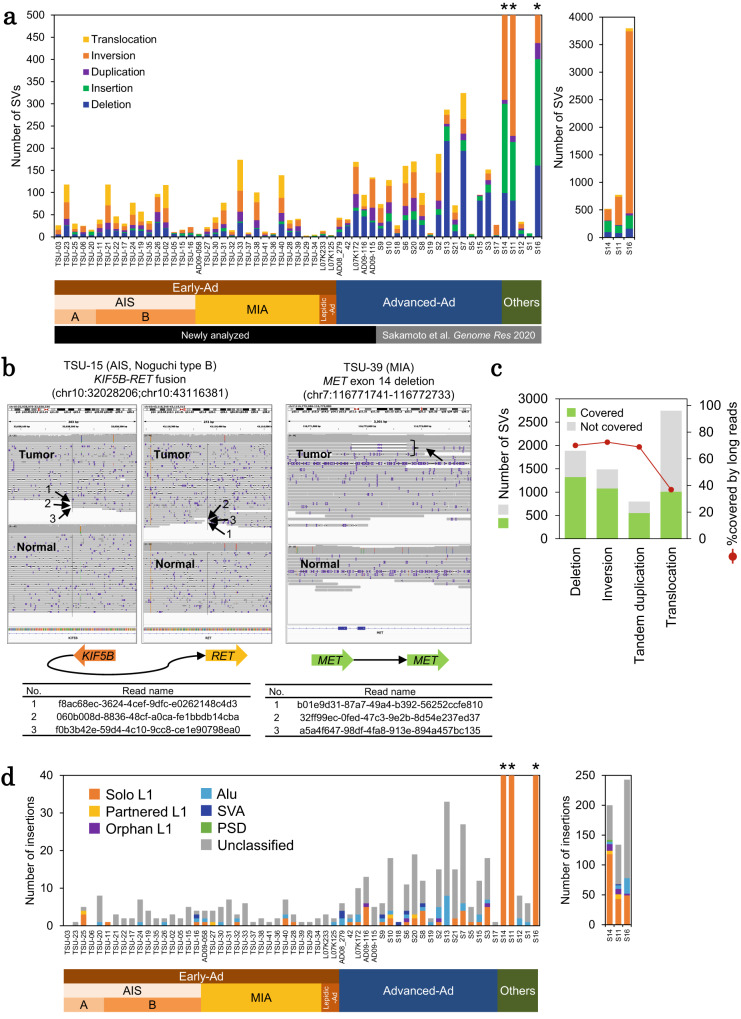
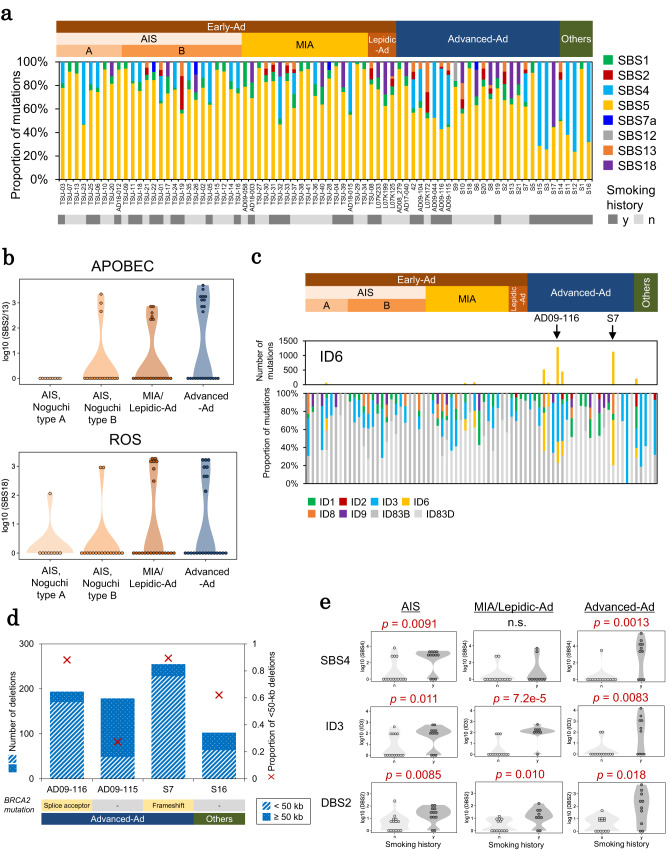
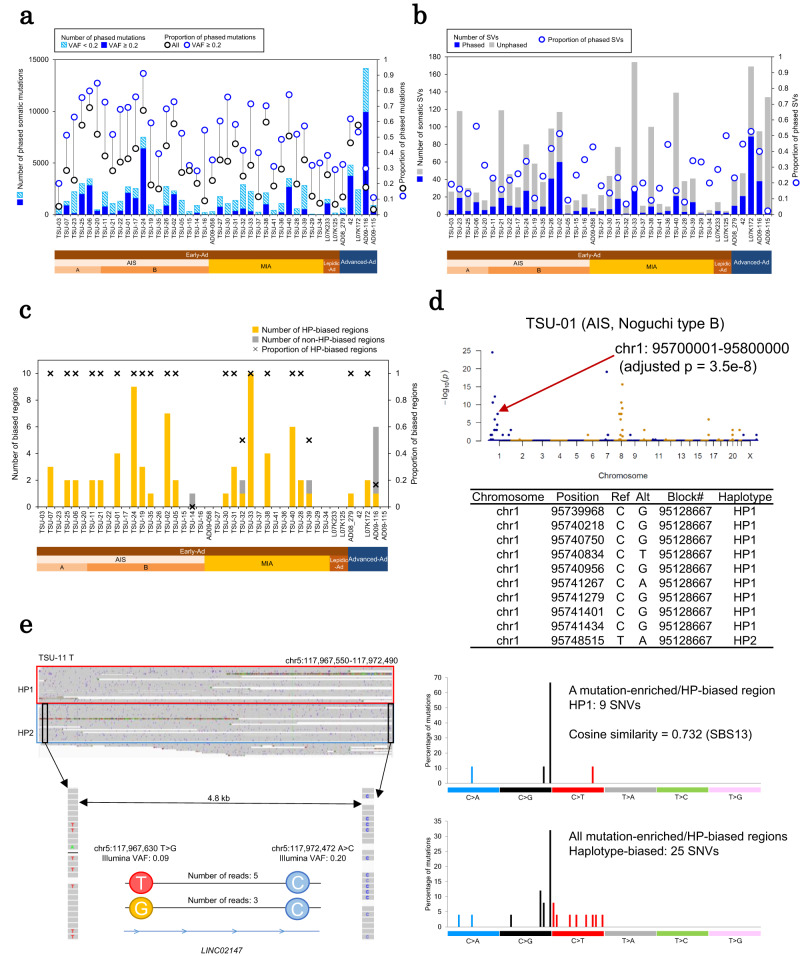
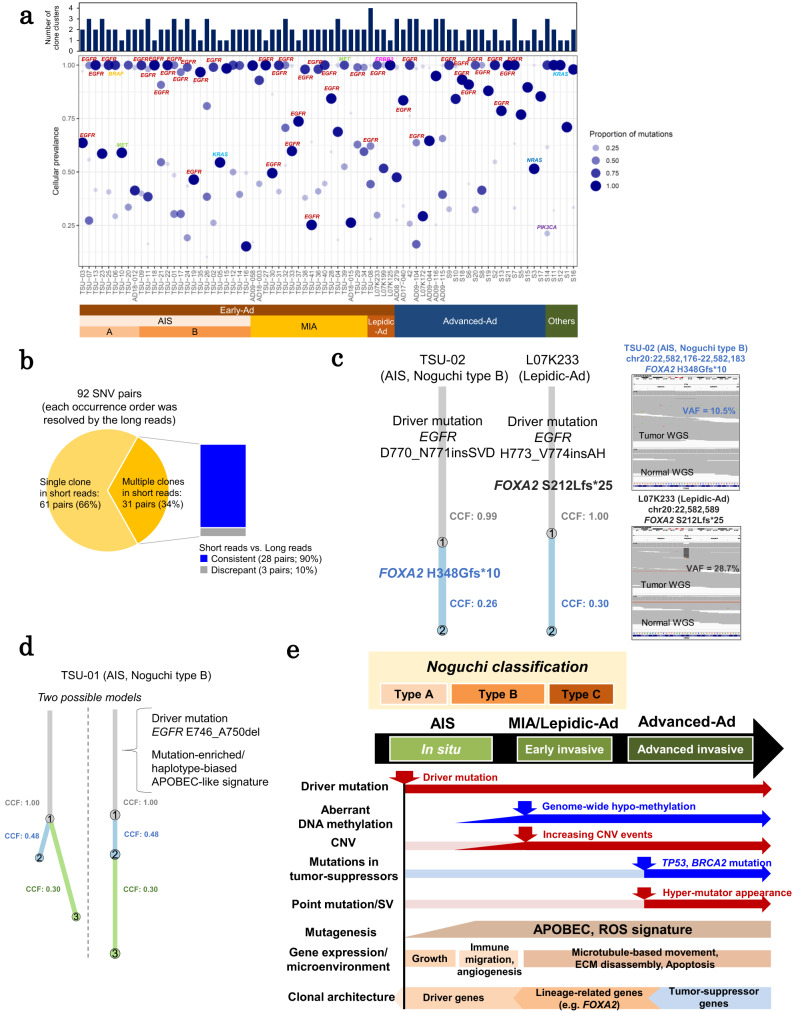
The mechanism underlying the development of tumors, particularly at early stages, still remains mostly elusive. Here, we report whole-genome long and short read sequencing analysis of 76 lung cancers, focusing on very early-stage lung adenocarcinomas such as adenocarcinoma in situ (AIS) and minimally invasive adenocarcinoma. The obtained data is further integrated with bulk and spatial transcriptomic data and epigenomic data. These analyses reveal key events in lung carcinogenesis. Minimal somatic mutations in pivotal driver mutations and essential proliferative factors are the only detectable somatic mutations in the very early-stage of AIS. These initial events are followed by copy number changes and global DNA hypomethylation. Particularly, drastic changes are initiated at the later AIS stage, i.e., in Noguchi type B tumors, wherein cancer cells are exposed to the surrounding microenvironment. This study sheds light on the pathogenesis of lung adenocarcinoma from integrated pathological and molecular viewpoints.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Kadota K, et al. Prognostic significance of adenocarcinoma in situ, minimally invasive adenocarcinoma, and nonmucinous lepidic predominant invasive adenocarcinoma of the lung in patients with stage I disease. Am. J. Surg. Pathol. 2014;38:448–460. doi: 10.1097/PAS.0000000000000134. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
