

PAX3-FOXO1 uses its activation domain to recruit CBP/P300 and shape RNA Pol2 cluster distribution
- PMID: 38102136
- PMCID: PMC10724205
- DOI: 10.1038/s41467-023-43780-4
PAX3-FOXO1 uses its activation domain to recruit CBP/P300 and shape RNA Pol2 cluster distribution
Abstract
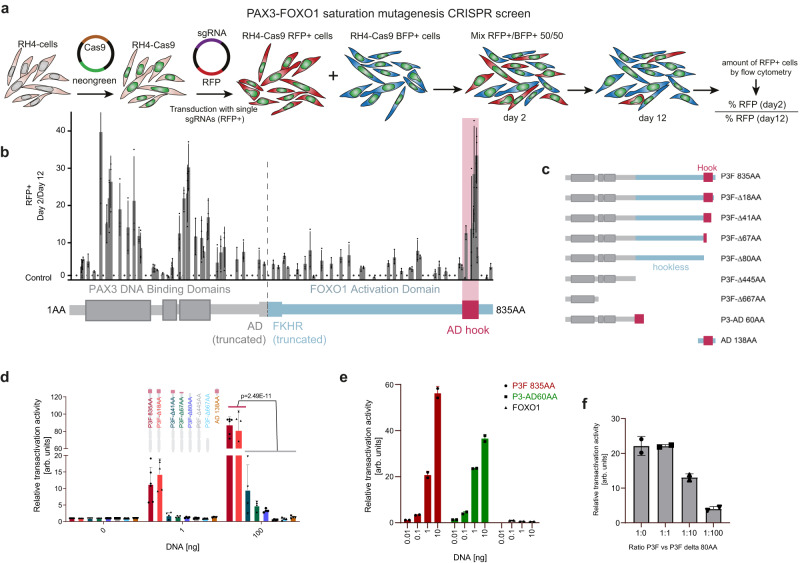
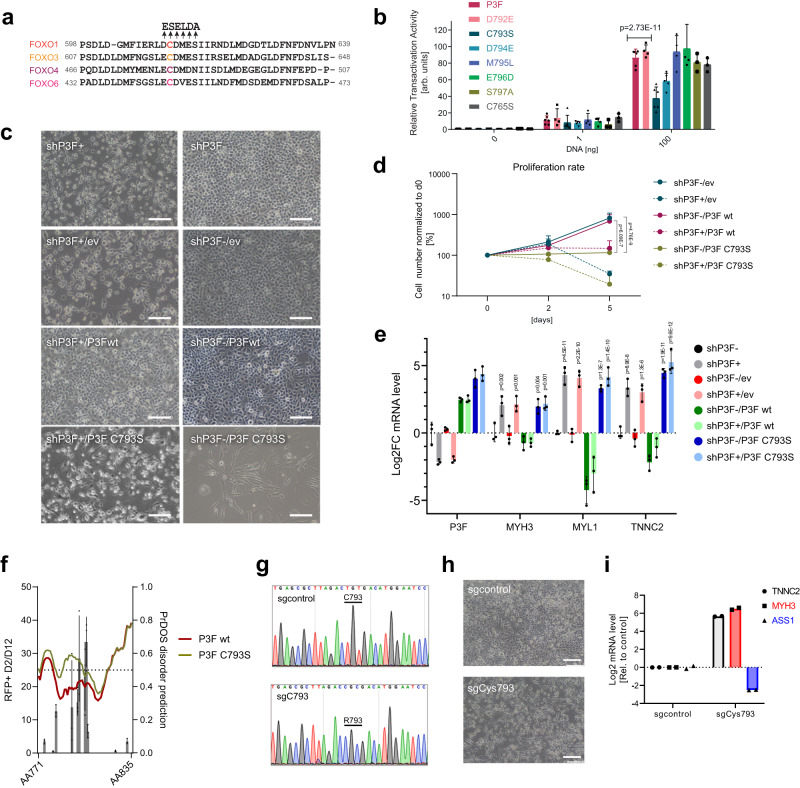
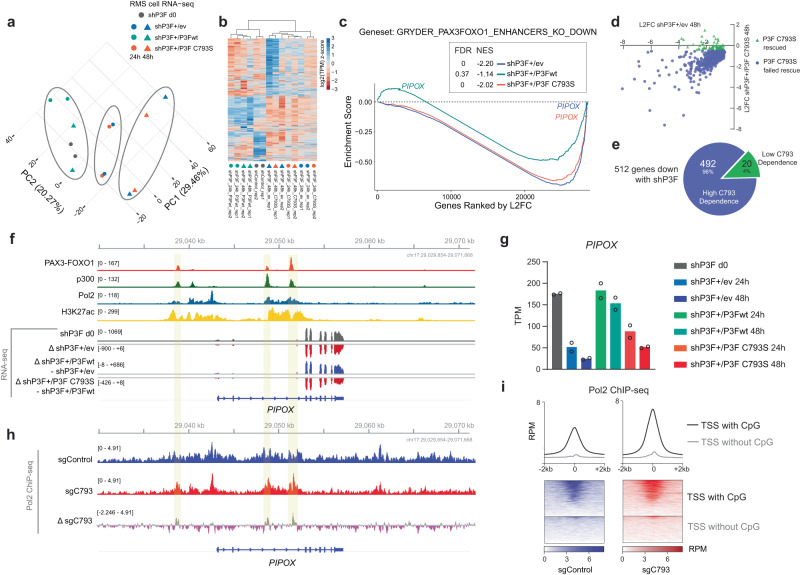
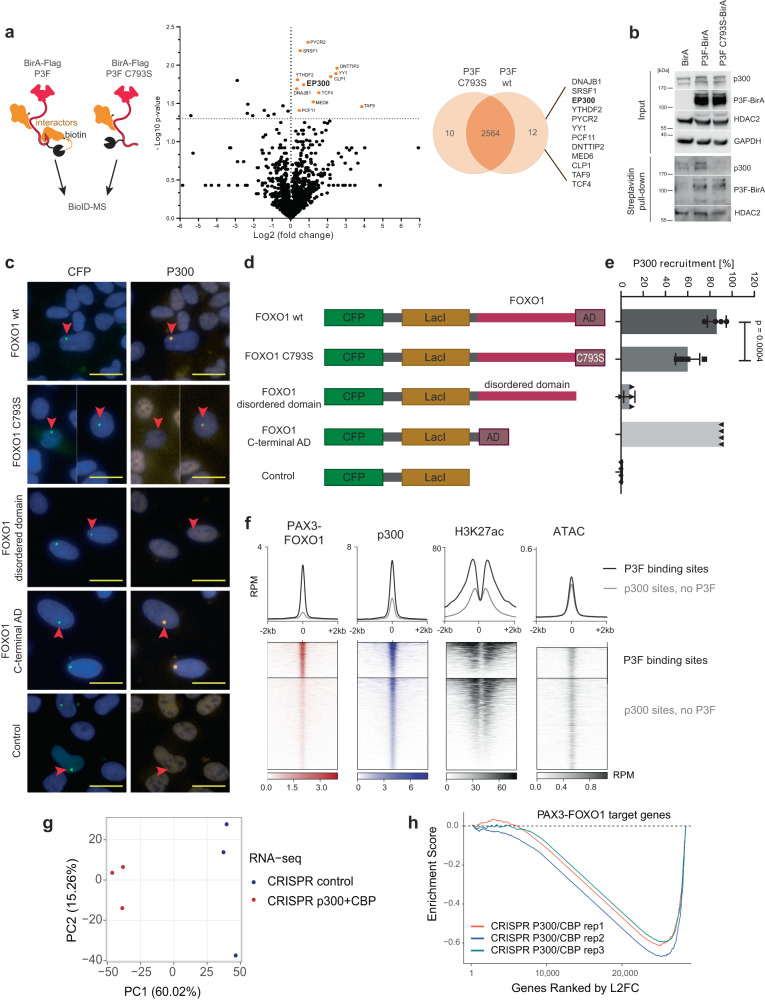
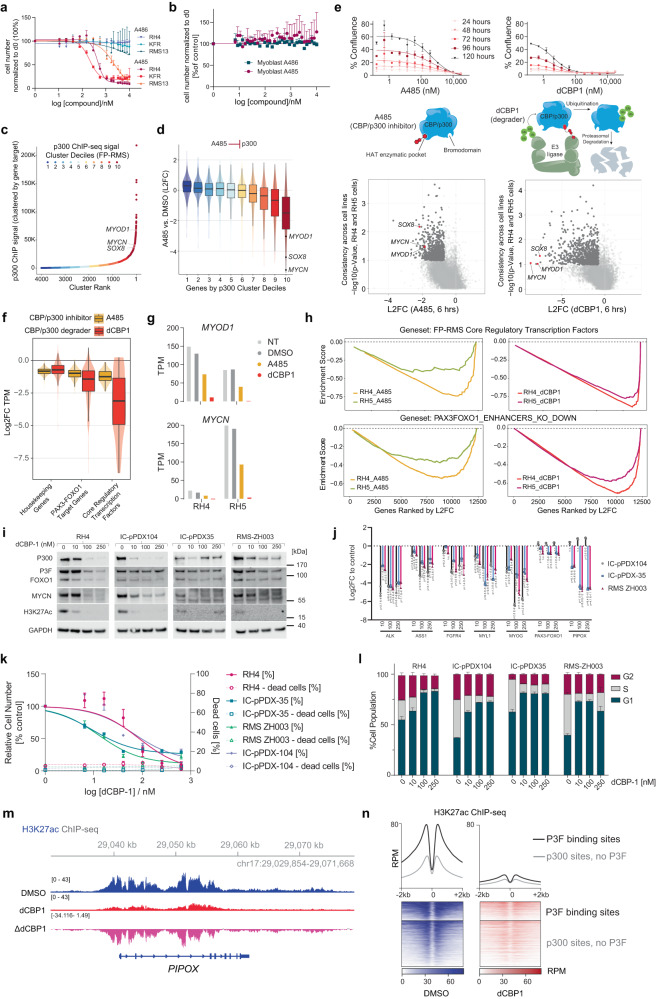
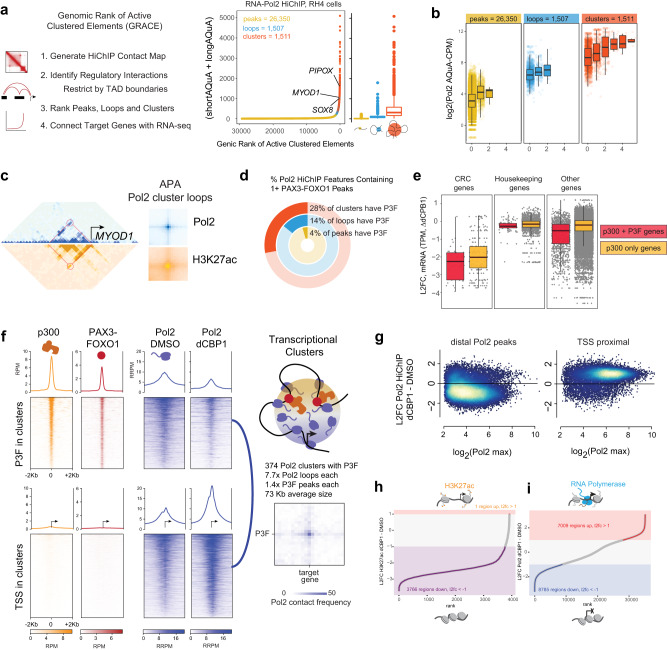
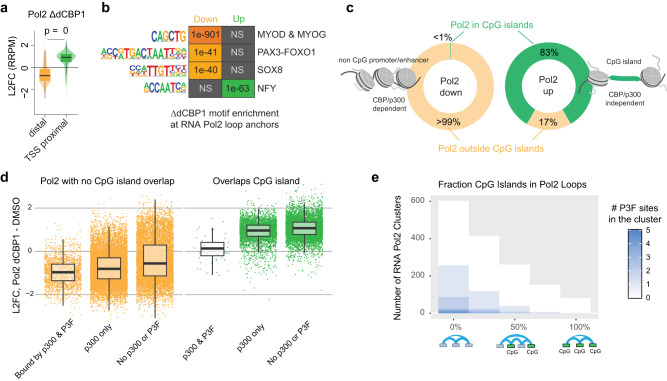
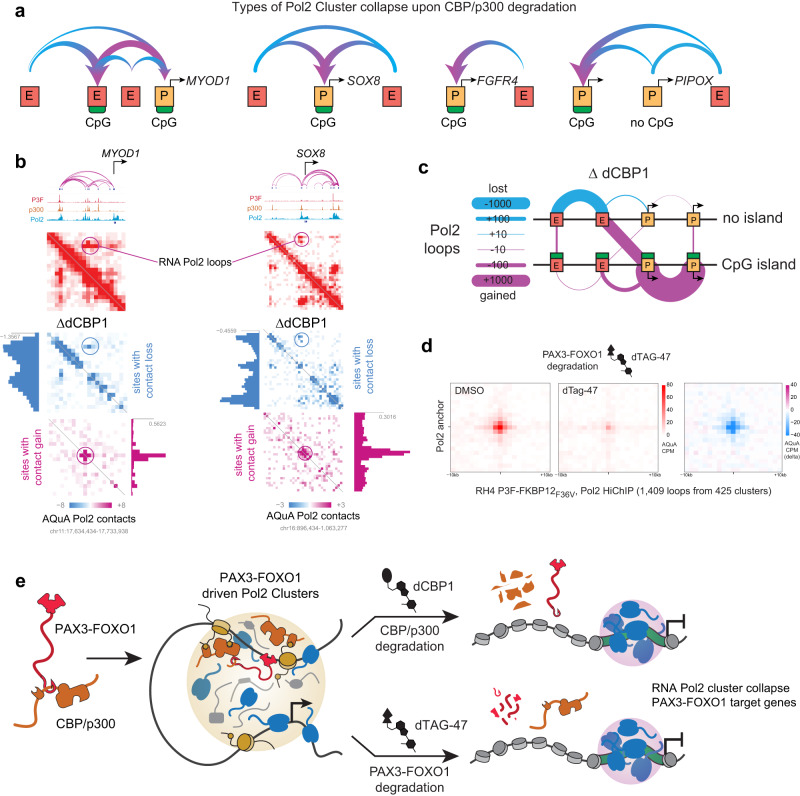
Activation of oncogenic gene expression from long-range enhancers is initiated by the assembly of DNA-binding transcription factors (TF), leading to recruitment of co-activators such as CBP/p300 to modify the local genomic context and facilitate RNA-Polymerase 2 (Pol2) binding. Yet, most TF-to-coactivator recruitment relationships remain unmapped. Here, studying the oncogenic fusion TF PAX3-FOXO1 (P3F) from alveolar rhabdomyosarcoma (aRMS), we show that a single cysteine in the activation domain (AD) of P3F is important for a small alpha helical coil that recruits CBP/p300 to chromatin. P3F driven transcription requires both this single cysteine and CBP/p300. Mutants of the cysteine reduce aRMS cell proliferation and induce cellular differentiation. Furthermore, we discover a profound dependence on CBP/p300 for clustering of Pol2 loops that connect P3F to its target genes. In the absence of CBP/p300, Pol2 long range enhancer loops collapse, Pol2 accumulates in CpG islands and fails to exit the gene body. These results reveal a potential novel axis for therapeutic interference with P3F in aRMS and clarify the molecular relationship of P3F and CBP/p300 in sustaining active Pol2 clusters essential for oncogenic transcription.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Gryder BE, et al. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017;7:884–899. doi: 10.1158/2159-8290.CD-16-1297. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
