

Diversity and dissemination of viruses in pathogenic protozoa
- PMID: 38102141
- PMCID: PMC10724245
- DOI: 10.1038/s41467-023-44085-2
Diversity and dissemination of viruses in pathogenic protozoa
Abstract
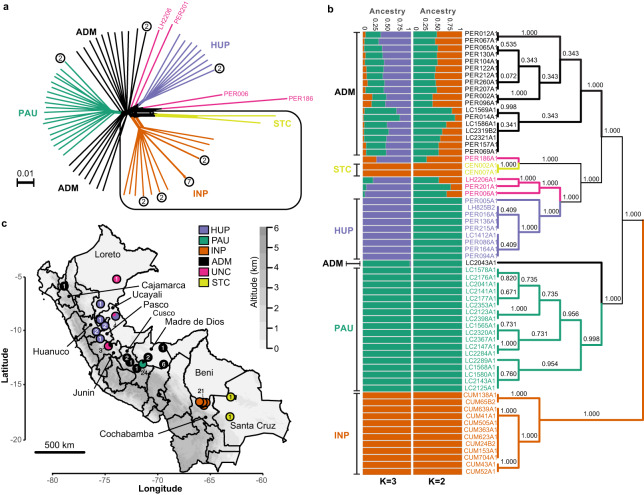
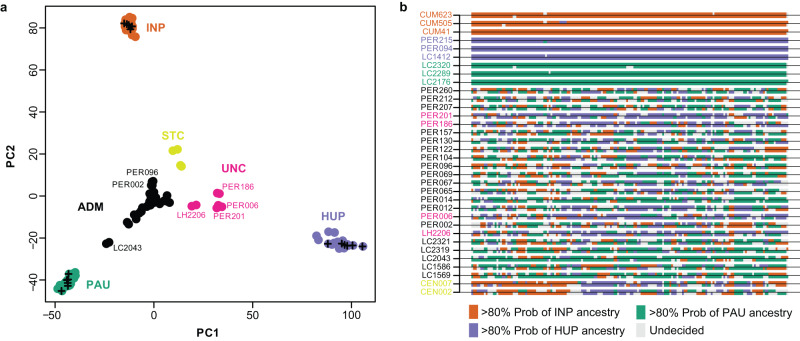
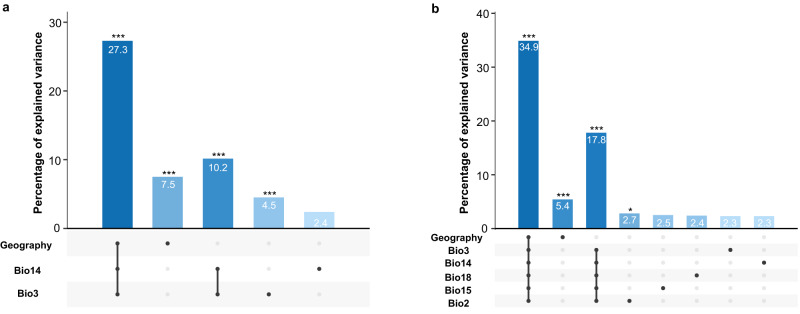
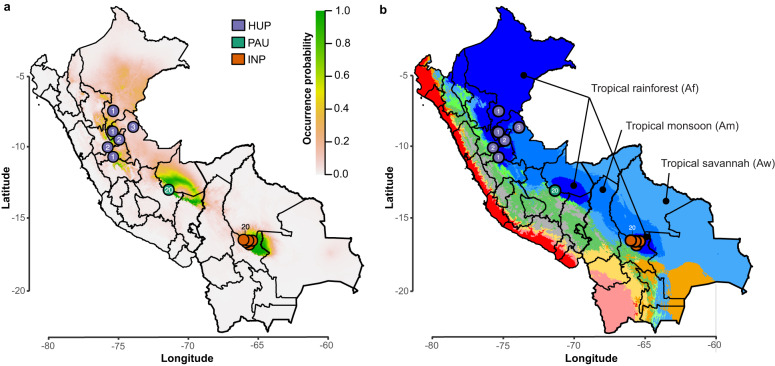
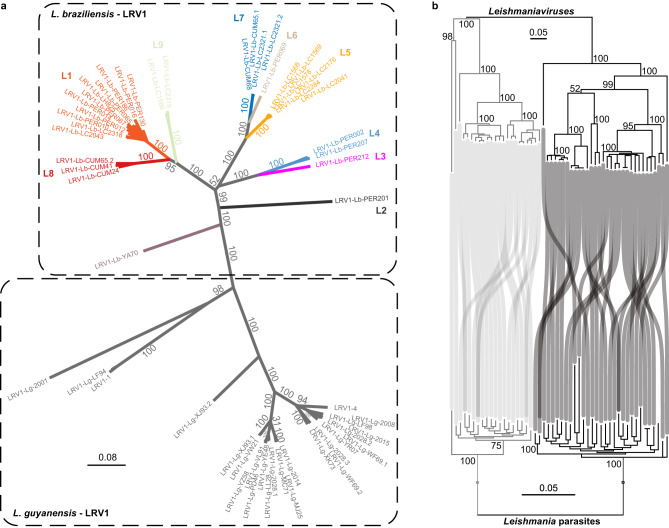
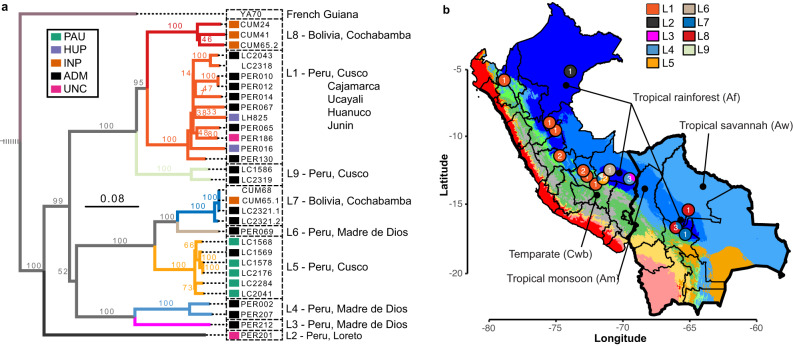
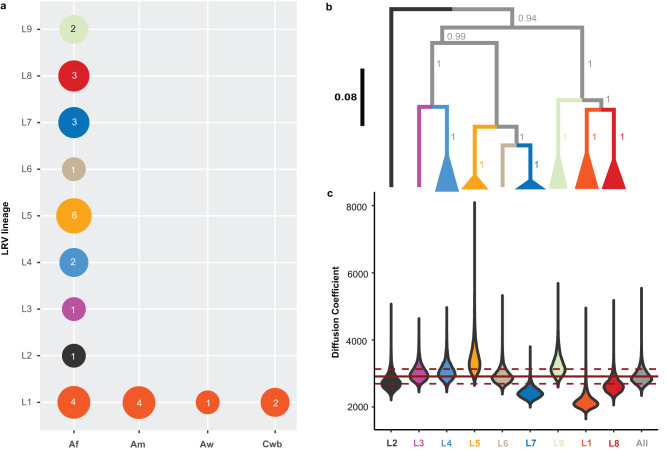
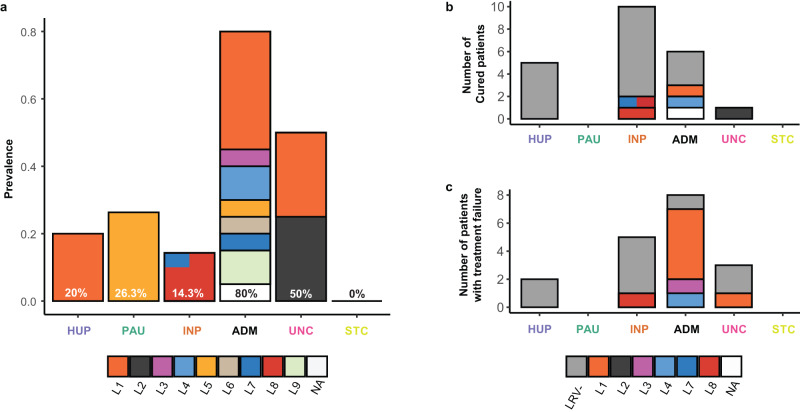
Viruses are the most abundant biological entities on Earth and play a significant role in the evolution of many organisms and ecosystems. In pathogenic protozoa, the presence of viruses has been linked to an increased risk of treatment failure and severe clinical outcome. Here, we studied the molecular epidemiology of the zoonotic disease cutaneous leishmaniasis in Peru and Bolivia through a joint evolutionary analysis of Leishmania braziliensis and their dsRNA Leishmania virus 1. We show that parasite populations circulate in tropical rainforests and are associated with single viral lineages that appear in low prevalence. In contrast, groups of hybrid parasites are geographically and ecologically more dispersed and associated with an increased prevalence, diversity and spread of viruses. Our results suggest that parasite gene flow and hybridization increased the frequency of parasite-virus symbioses, a process that may change the epidemiology of leishmaniasis in the region.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Update of
-
Parasite hybridization promotes spreading of endosymbiotic viruses.bioRxiv [Preprint]. 2023 Mar 25:2023.03.24.534103. doi: 10.1101/2023.03.24.534103. bioRxiv. 2023. Update in: Nat Commun. 2023 Dec 15;14(1):8343. doi: 10.1038/s41467-023-44085-2. PMID: 36993291 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
