

Crosstalk among mitophagy, pyroptosis, ferroptosis, and necroptosis in central nervous system injuries
- PMID: 38103229
- PMCID: PMC10960298
- DOI: 10.4103/1673-5374.389361
Crosstalk among mitophagy, pyroptosis, ferroptosis, and necroptosis in central nervous system injuries
Abstract
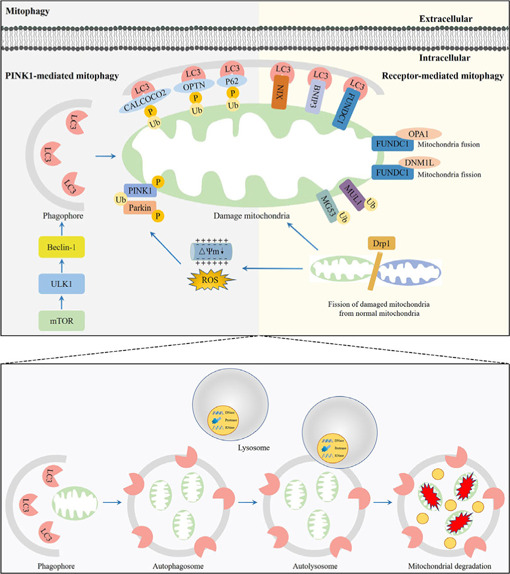
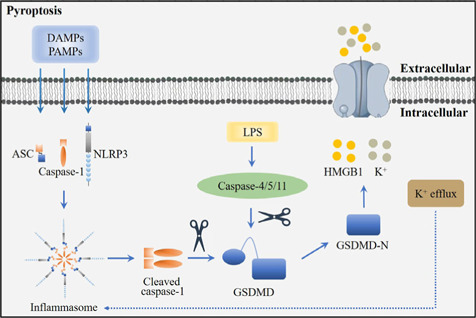
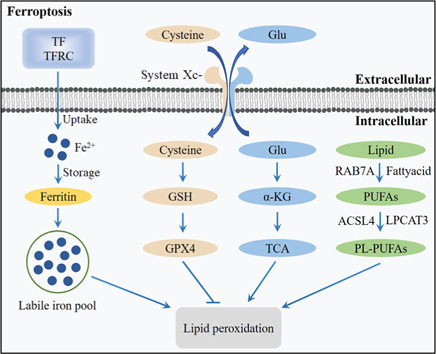
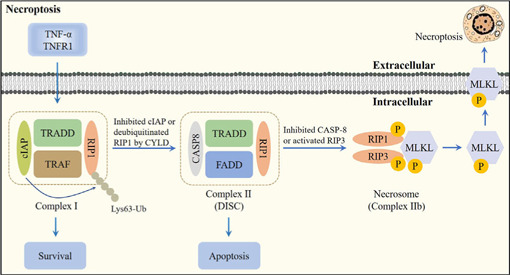
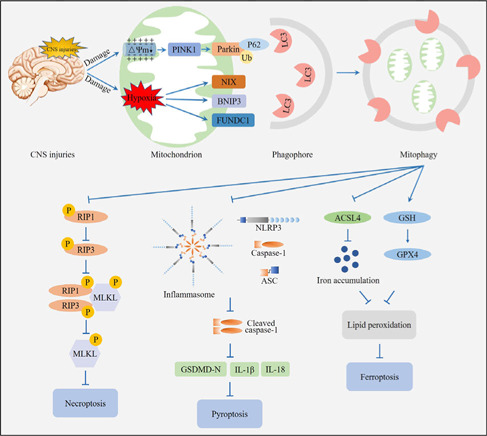
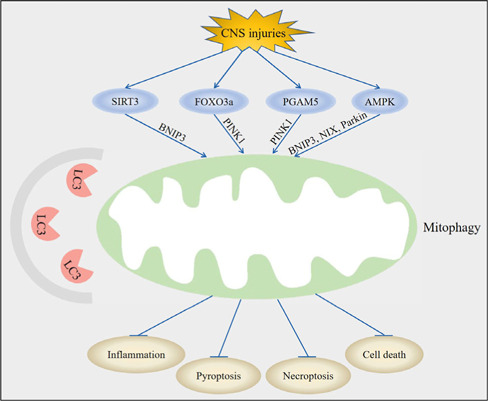
Central nervous system injuries have a high rate of resulting in disability and mortality; however, at present, effective treatments are lacking. Programmed cell death, which is a genetically determined form of active and ordered cell death with many types, has recently attracted increasing attention due to its functions in determining the fate of cell survival. A growing number of studies have suggested that programmed cell death is involved in central nervous system injuries and plays an important role in the progression of brain damage. In this review, we provide an overview of the role of programmed cell death in central nervous system injuries, including the pathways involved in mitophagy, pyroptosis, ferroptosis, and necroptosis, and the underlying mechanisms by which mitophagy regulates pyroptosis, ferroptosis, and necroptosis. We also discuss the new direction of therapeutic strategies targeting mitophagy for the treatment of central nervous system injuries, with the aim to determine the connection between programmed cell death and central nervous system injuries and to identify new therapies to modulate programmed cell death following central nervous system injury. In conclusion, based on these properties and effects, interventions targeting programmed cell death could be developed as potential therapeutic agents for central nervous system injury patients.
Copyright © 2024 Copyright: © 2024 Neural Regeneration Research.
Conflict of interest statement
Figures
Similar articles
-
Induction Mechanism of Ferroptosis, Necroptosis, and Pyroptosis: A Novel Therapeutic Target in Nervous System Diseases.Int J Mol Sci. 2023 Jun 14;24(12):10127. doi: 10.3390/ijms241210127. Int J Mol Sci. 2023. PMID: 37373274 Free PMC article. Review.
-
Ferroptosis, necroptosis, and pyroptosis in the occurrence and development of ovarian cancer.Front Immunol. 2022 Jul 25;13:920059. doi: 10.3389/fimmu.2022.920059. eCollection 2022. Front Immunol. 2022. PMID: 35958626 Free PMC article. Review.
-
Microglia programmed cell death in neurodegenerative diseases and CNS injury.Apoptosis. 2025 Feb;30(1-2):446-465. doi: 10.1007/s10495-024-02041-5. Epub 2024 Dec 10. Apoptosis. 2025. PMID: 39656359 Free PMC article. Review.
-
Targeting Programmed Cell Death in Acquired Sensorineural Hearing Loss: Ferroptosis, Necroptosis, and Pyroptosis.Neurosci Bull. 2025 Jun;41(6):1085-1102. doi: 10.1007/s12264-025-01370-y. Epub 2025 Apr 22. Neurosci Bull. 2025. PMID: 40261527 Free PMC article. Review.
-
The scheme, and regulative mechanism of pyroptosis, ferroptosis, and necroptosis in radiation injury.Int J Biol Sci. 2024 Mar 3;20(5):1871-1883. doi: 10.7150/ijbs.91112. eCollection 2024. Int J Biol Sci. 2024. PMID: 38481804 Free PMC article. Review.
Cited by
-
Forms of Non-Apoptotic Cell Death and Their Role in Gliomas-Presentation of the Current State of Knowledge.Biomedicines. 2024 Jul 11;12(7):1546. doi: 10.3390/biomedicines12071546. Biomedicines. 2024. PMID: 39062119 Free PMC article. Review.
-
Using ML techniques to predict extubation outcomes for patients with central nervous system injuries in the Yun-Gui Plateau.Sci Rep. 2025 May 22;15(1):17773. doi: 10.1038/s41598-025-98861-9. Sci Rep. 2025. PMID: 40404881 Free PMC article.
-
Insights on the crosstalk among different cell death mechanisms.Cell Death Discov. 2025 Feb 10;11(1):56. doi: 10.1038/s41420-025-02328-9. Cell Death Discov. 2025. PMID: 39929794 Free PMC article. Review.
-
BNIP3-mediated mitophagy attenuates hypoxic-ischemic brain damage in neonatal rats by inhibiting ferroptosis through P62-KEAP1-NRF2 pathway activation to maintain iron and redox homeostasis.Acta Pharmacol Sin. 2025 Jan;46(1):33-51. doi: 10.1038/s41401-024-01365-x. Epub 2024 Aug 23. Acta Pharmacol Sin. 2025. PMID: 39179868
-
Identifying the Potential Diagnostic Gene Biomarkers and Forecasting the Potential Therapeutic Agents for Advanced Diabetic Nephropathy Based on Pyroptosis and Ferroptosis.J Inflamm Res. 2024 Aug 29;17:5763-5779. doi: 10.2147/JIR.S467388. eCollection 2024. J Inflamm Res. 2024. PMID: 39224660 Free PMC article.
References
-
- Al Mamun A, Suchi SA, Aziz MA, Zaeem M, Munir F, Wu Y, Xiao J. Pyroptosis in acute pancreatitis and its therapeutic regulation. Apoptosis. 2022;27:465–481. - PubMed
-
- Appunni S, Gupta D, Rubens M, Ramamoorthy V, Singh HN, Swarup V. Deregulated protein kinases: friend and foe in ischemic stroke. Mol Neurobiol. 2021;58:6471–6489. - PubMed
-
- Chang H, Lin C, Li Z, Shen Y, Zhang G, Mao L, Ma C, Liu N, Lu H. T3 alleviates neuroinflammation and reduces early brain injury after subarachnoid haemorrhage by promoting mitophagy via PINK 1-parkin pathway. Exp Neurol. 2022;357:114175. - PubMed
-
- Chen J, Jin H, Xu H, Peng Y, Jie L, Xu D, Chen L, Li T, Fan L, He P, Ying G, Gu C, Wang C, Wang L, Chen G. The neuroprotective effects of necrostatin-1 on subarachnoid hemorrhage in rats are possibly mediated by preventing blood-brain barrier disruption and RIP3-mediated necroptosis. Cell Transplant. 2019;28:1358–1372. - PMC - PubMed
LinkOut - more resources
Full Text Sources
