

Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies
- PMID: 38105263
- PMCID: PMC10725898
- DOI: 10.1038/s41392-023-01705-z
Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies
Abstract
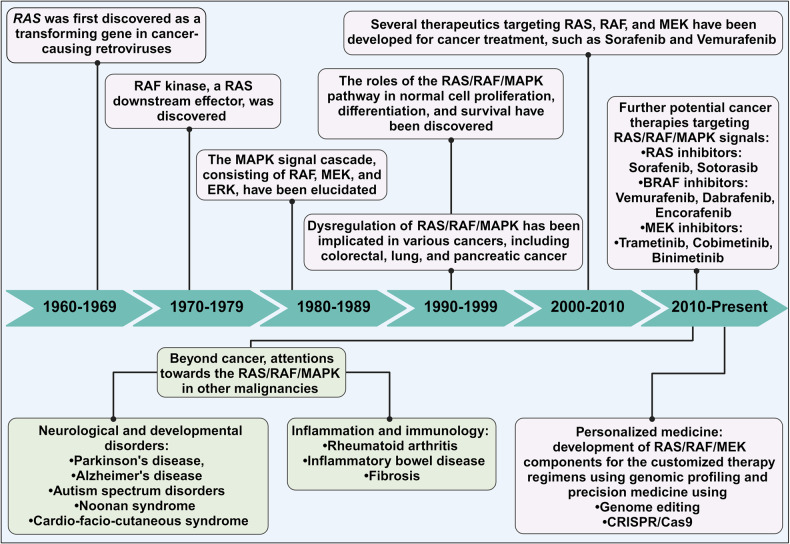
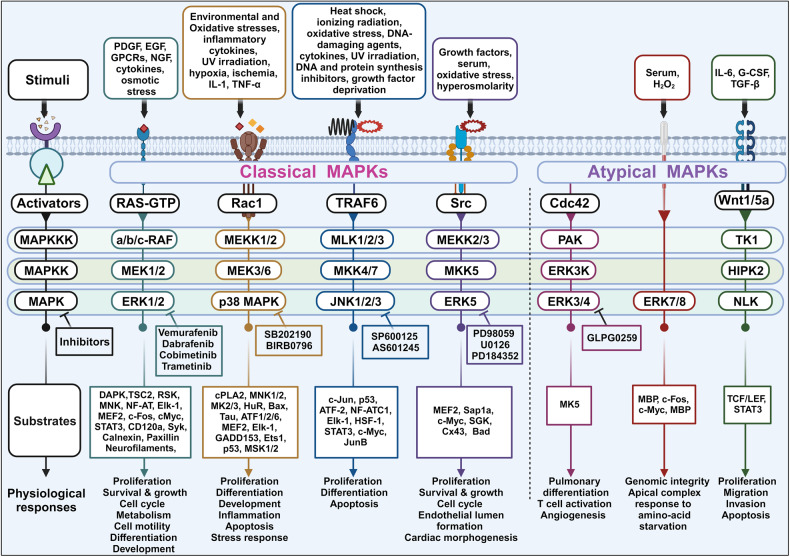
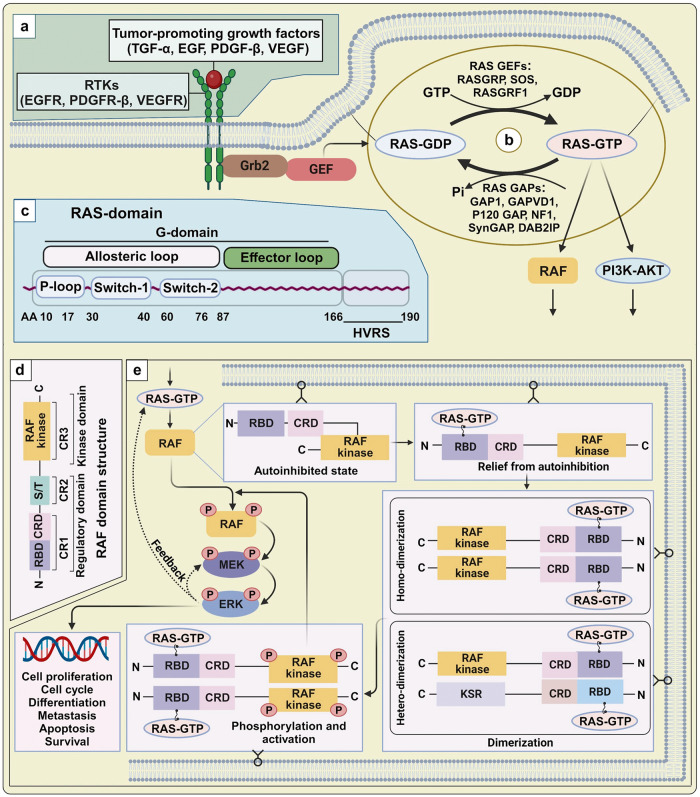
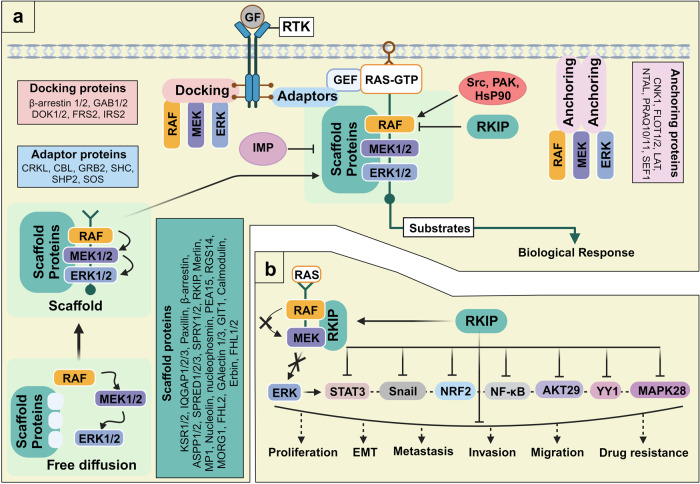
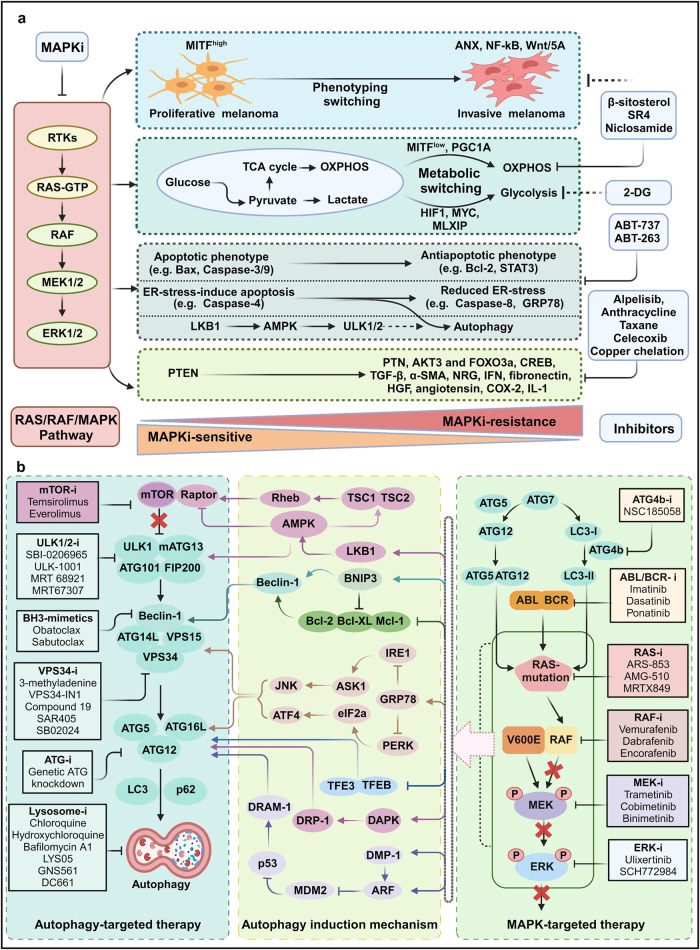
Metastatic dissemination of solid tumors, a leading cause of cancer-related mortality, underscores the urgent need for enhanced insights into the molecular and cellular mechanisms underlying metastasis, chemoresistance, and the mechanistic backgrounds of individuals whose cancers are prone to migration. The most prevalent signaling cascade governed by multi-kinase inhibitors is the mitogen-activated protein kinase (MAPK) pathway, encompassing the RAS-RAF-MAPK kinase (MEK)-extracellular signal-related kinase (ERK) pathway. RAF kinase is a primary mediator of the MAPK pathway, responsible for the sequential activation of downstream targets, such as MEK and the transcription factor ERK, which control numerous cellular and physiological processes, including organism development, cell cycle control, cell proliferation and differentiation, cell survival, and death. Defects in this signaling cascade are associated with diseases such as cancer. RAF inhibitors (RAFi) combined with MEK blockers represent an FDA-approved therapeutic strategy for numerous RAF-mutant cancers, including melanoma, non-small cell lung carcinoma, and thyroid cancer. However, the development of therapy resistance by cancer cells remains an important barrier. Autophagy, an intracellular lysosome-dependent catabolic recycling process, plays a critical role in the development of RAFi resistance in cancer. Thus, targeting RAF and autophagy could be novel treatment strategies for RAF-mutant cancers. In this review, we delve deeper into the mechanistic insights surrounding RAF kinase signaling in tumorigenesis and RAFi-resistance. Furthermore, we explore and discuss the ongoing development of next-generation RAF inhibitors with enhanced therapeutic profiles. Additionally, this review sheds light on the functional interplay between RAF-targeted therapies and autophagy in cancer.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
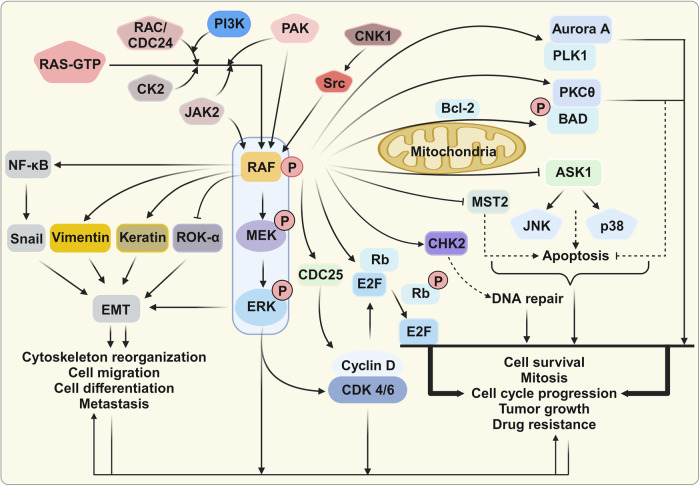
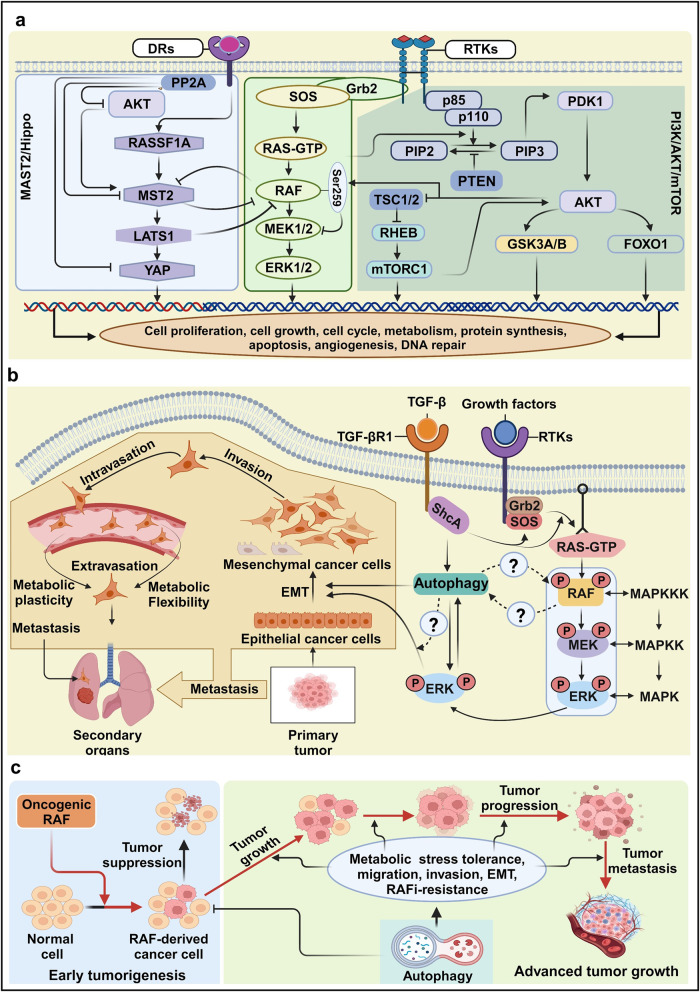
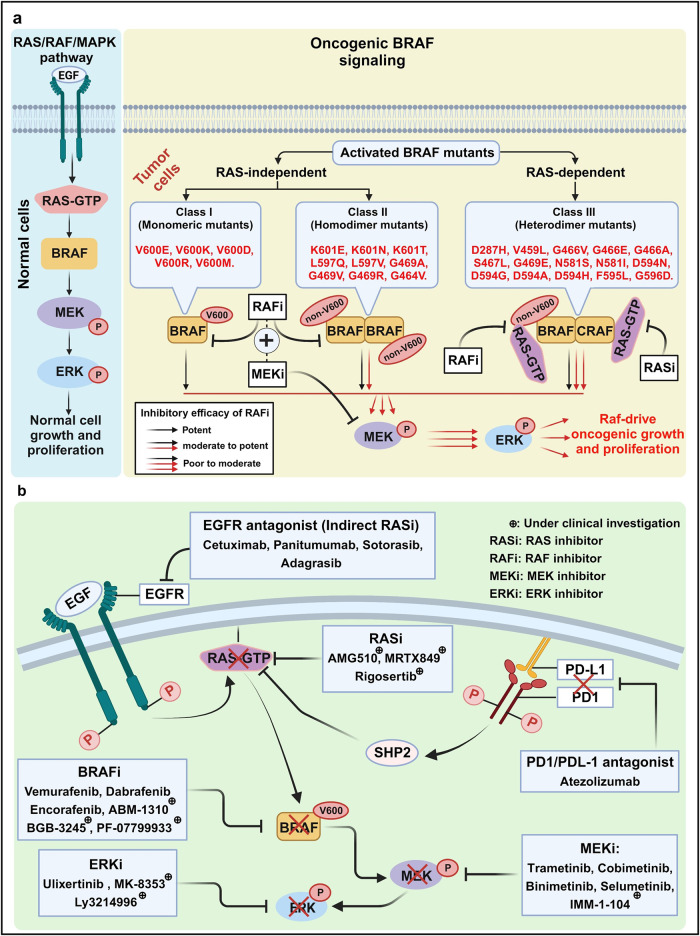
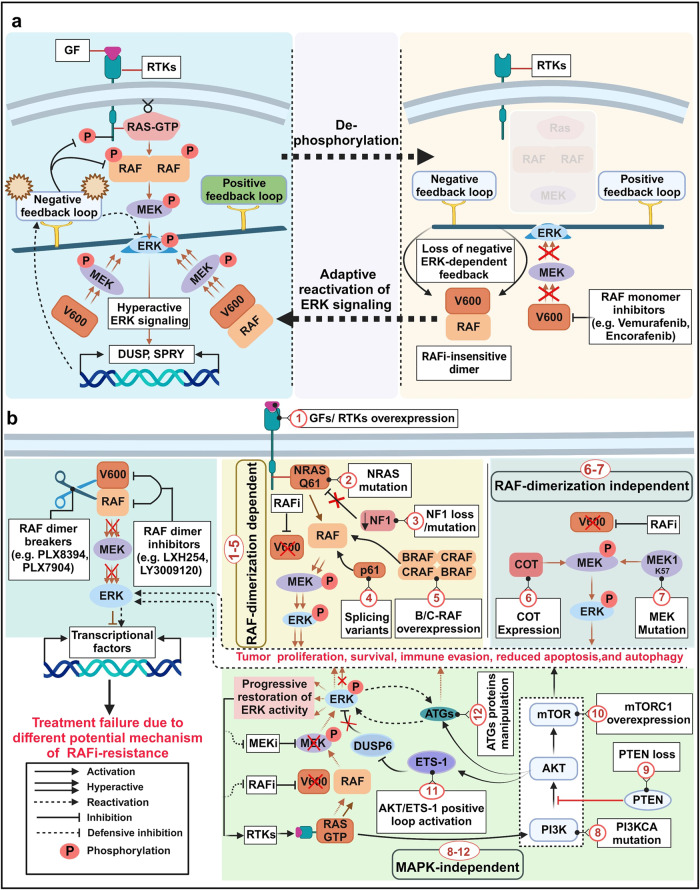
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
