

Conditional knockout of AIM2 in microglia ameliorates synaptic plasticity and spatial memory deficits in a mouse model of Alzheimer's disease
- PMID: 38105588
- PMCID: PMC11163192
- DOI: 10.1111/cns.14555
Conditional knockout of AIM2 in microglia ameliorates synaptic plasticity and spatial memory deficits in a mouse model of Alzheimer's disease
Abstract
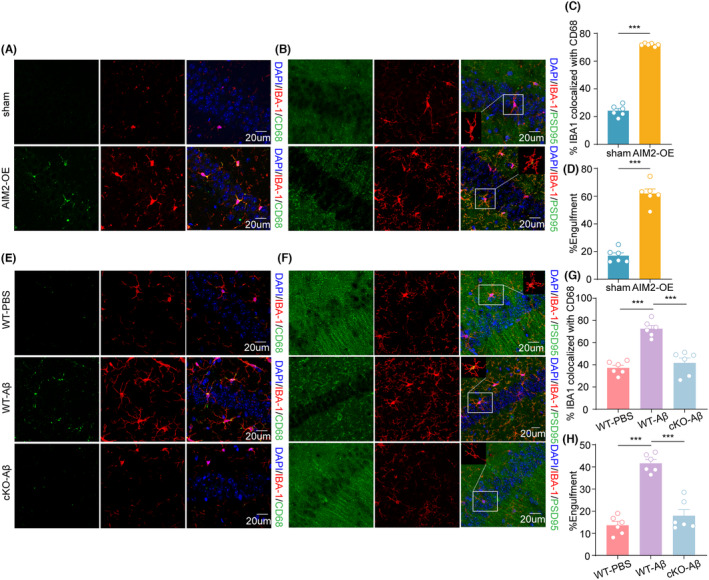
Aims: Synaptic dysfunction is a hallmark pathology of Alzheimer's disease (AD) and is strongly associated with cognitive impairment. Abnormal phagocytosis by the microglia is one of the main causes of synapse loss in AD. Previous studies have shown that the absence of melanoma 2 (AIM2) inflammasome activity is increased in the hippocampus of APP/PS1 mice, but the role of AIM2 in AD remains unclear.
Methods: Injection of Aβ1-42 into the bilateral hippocampal CA1 was used to mimic an AD mouse model (AD mice). C57BL/6 mice injected with AIM2 overexpression lentivirus and conditional knockout of microglial AIM2 mice were used to confirm the function of AIM2 in AD. Cognitive functions were assessed with novel object recognition and Morris water maze tests. The protein and mRNA expression levels were evaluated by western blotting, immunofluorescence staining, and qRT-PCR. Synaptic structure and function were detected by Golgi staining and electrophysiology.
Results: The expression level of AIM2 was increased in AD mice, and overexpression of AIM2 induced synaptic and cognitive impairments in C57BL/6 mice, similar to AD mice. Elevated expression levels of AIM2 occurred in microglia in AD mice. Conditional knockout of microglial AIM2 rescued cognitive and synaptic dysfunction in AD mice. Excessive microglial phagocytosis activity of synapses was decreased after knockout of microglial AIM2, which was associated with inhibiting complement activation.
Conclusion: Our results demonstrated that microglial AIM2 plays a critical role in regulating synaptic plasticity and memory deficits associated with AD, providing a new direction for developing novel preventative and therapeutic interventions for this disease.
Keywords: AIM2; Alzheimer's disease; complement; microglia; synaptic loss.
© 2023 The Authors. CNS Neuroscience & Therapeutics published by John Wiley & Sons Ltd.
Conflict of interest statement
Xu, Yun is an Editorial Board member of CNS Neuroscience and Therapeutics and a coauthor of this article. To minimize bias, they were excluded from all editorial decision‐making related to the acceptance of this article for publication.
Figures





Similar articles
-
Cognitive improvement effects of PF-04957325, a phosphodiesterase-8 inhibitor, in mouse models of Alzheimer's disease via modulating neuroinflammation.Int J Neuropsychopharmacol. 2025 May 9;28(5):pyaf028. doi: 10.1093/ijnp/pyaf028. Int J Neuropsychopharmacol. 2025. PMID: 40312965 Free PMC article.
-
Inhibition of FAM19A5 reverses synaptic loss and cognitive decline in mouse models of Alzheimer's disease.Alzheimers Res Ther. 2025 Jul 21;17(1):168. doi: 10.1186/s13195-025-01813-8. Alzheimers Res Ther. 2025. PMID: 40691577 Free PMC article. Clinical Trial.
-
Mitigation of synaptic and memory impairments via F-actin stabilization in Alzheimer's disease.Alzheimers Res Ther. 2024 Sep 7;16(1):200. doi: 10.1186/s13195-024-01558-w. Alzheimers Res Ther. 2024. PMID: 39244567 Free PMC article.
-
Selegiline for Alzheimer's disease.Cochrane Database Syst Rev. 2003;(1):CD000442. doi: 10.1002/14651858.CD000442. Cochrane Database Syst Rev. 2003. PMID: 12535396
-
Microglial activation as a hallmark of neuroinflammation in Alzheimer's disease.Metab Brain Dis. 2025 May 17;40(5):207. doi: 10.1007/s11011-025-01631-9. Metab Brain Dis. 2025. PMID: 40381069 Review.
Cited by
-
The roles of AIM2 in neurodegenerative diseases: insights and therapeutic implications.Front Immunol. 2024 Jul 15;15:1441385. doi: 10.3389/fimmu.2024.1441385. eCollection 2024. Front Immunol. 2024. PMID: 39076969 Free PMC article. Review.
-
Bibliometric analysis of pathological mechanisms in Alzheimer's disease: Applications based on mouse models.J Alzheimers Dis Rep. 2025 Aug 13;9:25424823251367046. doi: 10.1177/25424823251367046. eCollection 2025 Jan-Dec. J Alzheimers Dis Rep. 2025. PMID: 40822130 Free PMC article. Review.
-
AIM2-Driven Inflammation in Periodontitis: Mechanisms and Systemic Implications.J Inflamm Res. 2025 May 30;18:6983-6997. doi: 10.2147/JIR.S505907. eCollection 2025. J Inflamm Res. 2025. PMID: 40463858 Free PMC article.
-
MeCP2 Lactylation Protects against Ischemic Brain Injury by Transcriptionally Regulating Neuronal Apoptosis.Adv Sci (Weinh). 2025 Jun;12(21):e2415309. doi: 10.1002/advs.202415309. Epub 2025 Apr 24. Adv Sci (Weinh). 2025. PMID: 40271828 Free PMC article.
-
Inflammasomes in neurodegenerative diseases.Transl Neurodegener. 2024 Dec 23;13(1):65. doi: 10.1186/s40035-024-00459-0. Transl Neurodegener. 2024. PMID: 39710713 Free PMC article. Review.
References
-
- Peng L, Bestard‐Lorigados I, Song W. The synapse as a treatment avenue for Alzheimer's disease. Mol Psychiatry. 2022;27(7):2940‐2949. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
