

This is a preprint.
Multiplexed RNA-FISH-guided Laser Capture Microdissection RNA Sequencing Improves Breast Cancer Molecular Subtyping, Prognostic Classification, and Predicts Response to Antibody Drug Conjugates
- PMID: 38105959
- PMCID: PMC10723508
- DOI: 10.1101/2023.12.05.23299341
Multiplexed RNA-FISH-guided Laser Capture Microdissection RNA Sequencing Improves Breast Cancer Molecular Subtyping, Prognostic Classification, and Predicts Response to Antibody Drug Conjugates
Update in
-
The spatially informed mFISHseq assay resolves biomarker discordance and predicts treatment response in breast cancer.Nat Commun. 2025 Jan 2;16(1):226. doi: 10.1038/s41467-024-55583-2. Nat Commun. 2025. PMID: 39747865 Free PMC article. Clinical Trial.
Abstract
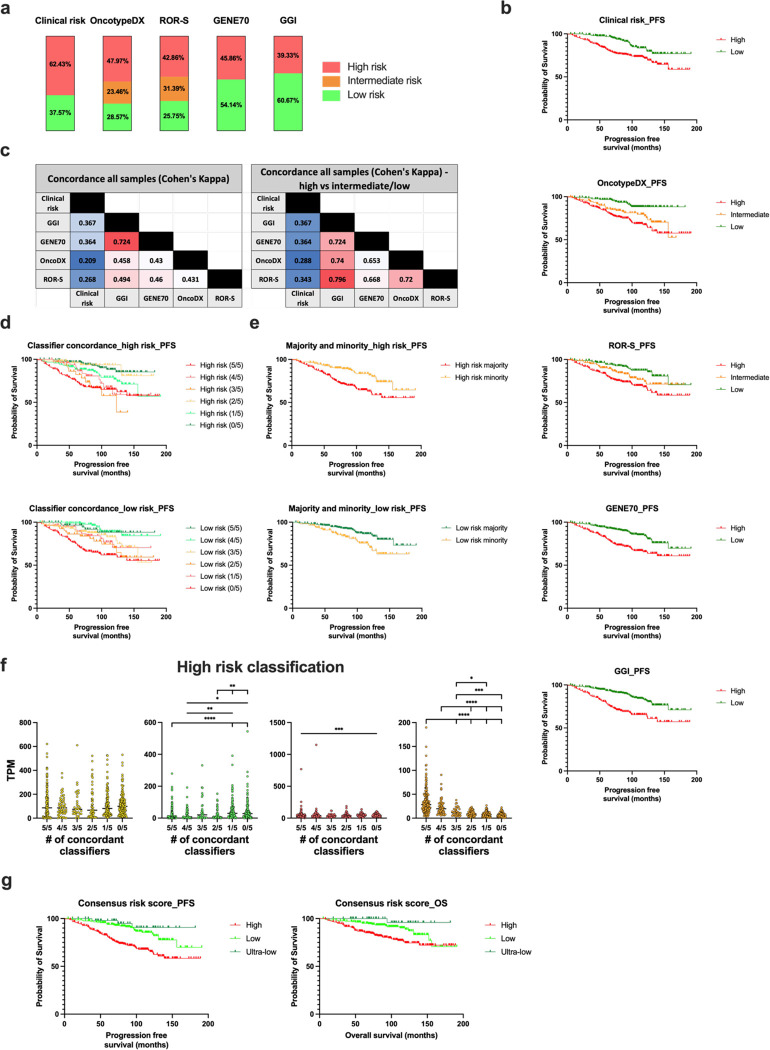
On a retrospective cohort of 1,082 FFPE breast tumors, we demonstrated the analytical validity of a test using multiplexed RNA-FISH-guided laser capture microdissection (LCM) coupled with RNA-sequencing (mFISHseq), which showed 93% accuracy compared to immunohistochemistry. The combination of these technologies makes strides in i) precisely assessing tumor heterogeneity, ii) obtaining pure tumor samples using LCM to ensure accurate biomarker expression and multigene testing, and iii) providing thorough and granular data from whole transcriptome profiling. We also constructed a 293-gene intrinsic subtype classifier that performed equivalent to the research based PAM50 and AIMS classifiers. By combining three molecular classifiers for consensus subtyping, mFISHseq alleviated single sample discordance, provided near perfect concordance with other classifiers (κ > 0.85), and reclassified 30% of samples into different subtypes with prognostic implications. We also use a consensus approach to combine information from 4 multigene prognostic classifiers and clinical risk to characterize high, low, and ultra-low risk patients that relapse early (< 5 years), late (> 10 years), and rarely, respectively. Lastly, to identify potential patient subpopulations that may be responsive to treatments like antibody drug-conjugates (ADC), we curated a list of 92 genes and 110 gene signatures to interrogate their association with molecular subtype and overall survival. Many genes and gene signatures related to ADC processing (e.g., antigen/payload targets, endocytosis, and lysosome activity) were independent predictors of overall survival in multivariate Cox regression models, thus highlighting potential ADC treatment-responsive subgroups. To test this hypothesis, we constructed a unique 19-feature classifier using multivariate logistic regression with elastic net that predicted response to trastuzumab emtansine (T-DM1; AUC = 0.96) better than either ERBB2 mRNA or Her2 IHC alone in the T-DM1 arm of the I-SPY2 trial. This test was deployed in a research-use only format on 26 patients and revealed clinical insights into patient selection for novel therapies like ADCs and immunotherapies and de-escalation of adjuvant chemotherapy.
Keywords: RNA-FISH; RNA-sequencing; antibody drug-conjugates; breast cancer; laser capture microdissection; spatial biology.
Conflict of interest statement
Competing interests All authors have completed the ICMJE uniform disclosure form at www.icmje.org/coi_disclosure.pdf and declared the following: E.D.P., B.H., N.Val., N.B., D.G., S.G., H.I., T.O., S.B., J.B., K.B., D.D., Z.K., M.K., D.L., V.Mam., V.Man., N.Voj., and P.Č. are current, or former employees of MultiplexDX, a biotechnology company that is developing a lab developed diagnostic test called Multiplex8+ (https://www.multiplexdx.com/products/multiplex-eight-plus), which is based on the research presented in the manuscript. P.Č. and E.D.P. are inventors and MultiplexDX, s.r.o is the assignee on patent applications that were filed in relation to the technology and research outlined in the manuscript. T.T. and F.P. are members of the Scientific Advisory board at MultiplexDX. All other authors declare no competing interests.
Figures









References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous