

This is a preprint.
An evolution-based framework for describing human gut bacteria
- PMID: 38105970
- PMCID: PMC10723311
- DOI: 10.1101/2023.12.04.569969
An evolution-based framework for describing human gut bacteria
Abstract
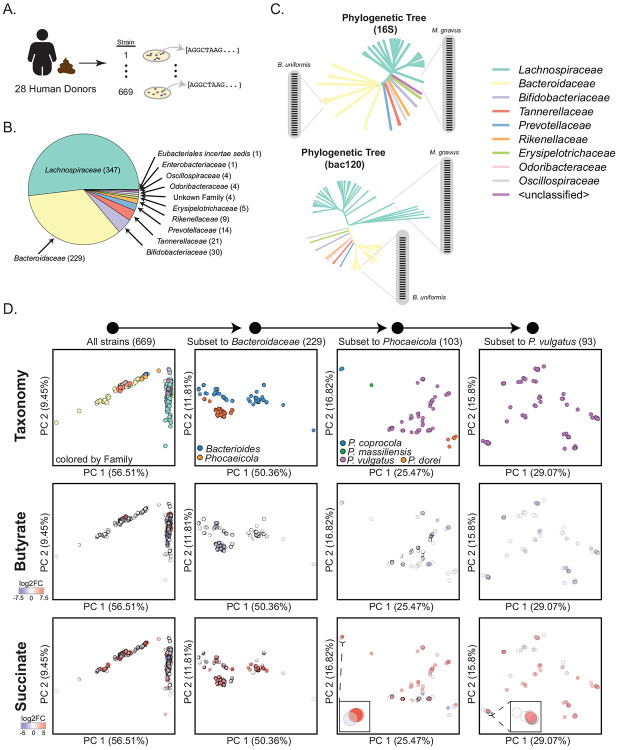
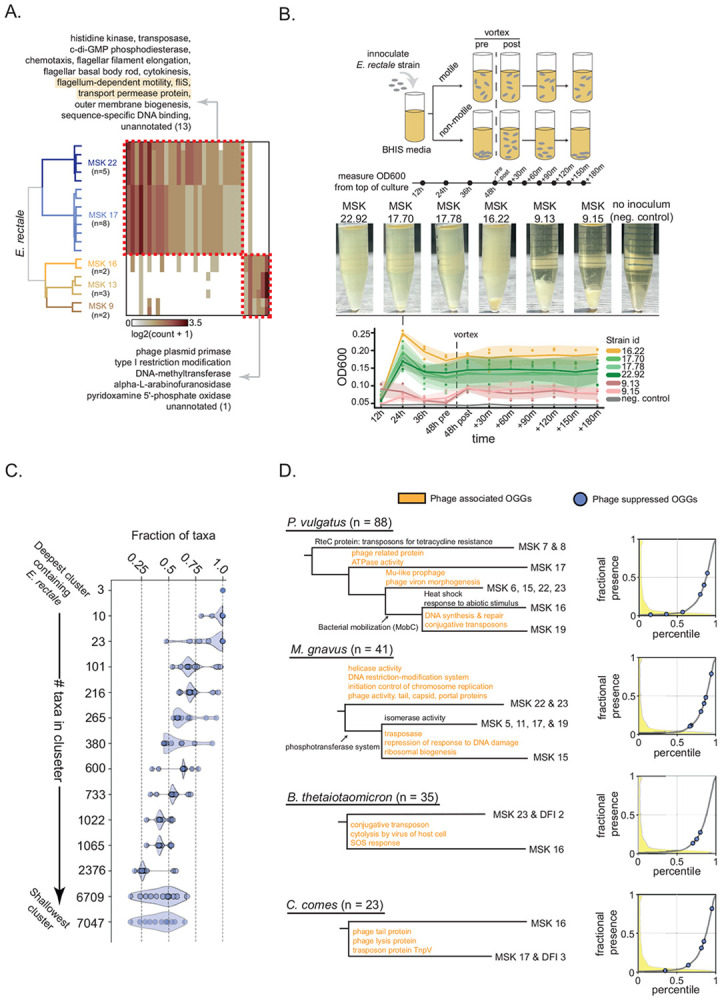
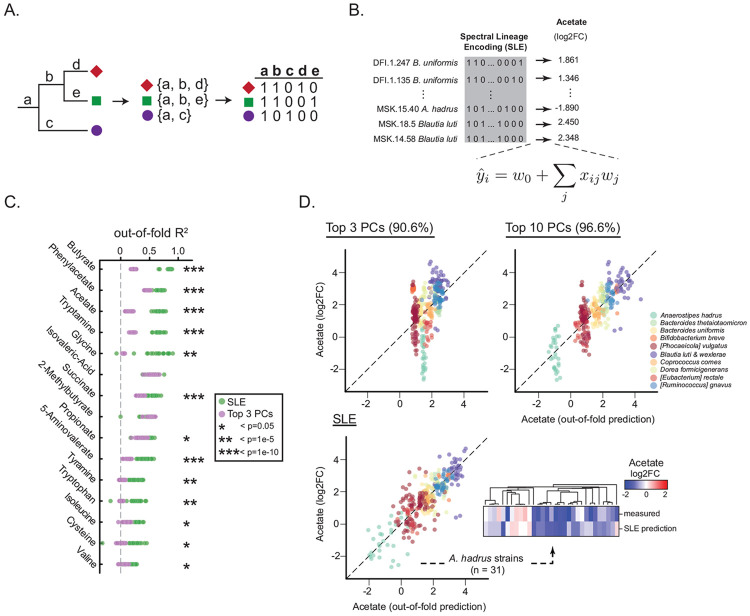
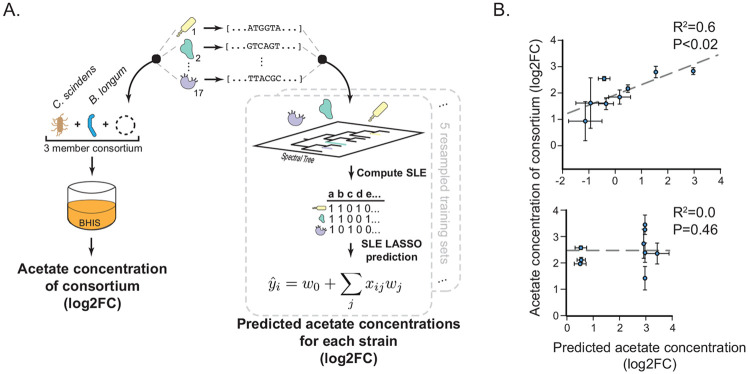
The human gut microbiome contains many bacterial strains of the same species ('strain-level variants'). Describing strains in a biologically meaningful way rather than purely taxonomically is an important goal but challenging due to the genetic complexity of strain-level variation. Here, we measured patterns of co-evolution across >7,000 strains spanning the bacterial tree-of-life. Using these patterns as a prior for studying hundreds of gut commensal strains that we isolated, sequenced, and metabolically profiled revealed widespread structure beneath the phylogenetic level of species. Defining strains by their co-evolutionary signatures enabled predicting their metabolic phenotypes and engineering consortia from strain genome content alone. Our findings demonstrate a biologically relevant organization to strain-level variation and motivate a new schema for describing bacterial strains based on their evolutionary history.
Figures

References
-
- Sunagawa S., Acinas S. G., Bork P., Bowler C., Tara Oceans Coordinators, Eveillard D., Gorsky G., Guidi L., Iudicone D., Karsenti E., Lombard F., Ogata H., Pesant S., Sullivan M. B., Wincker P., de Vargas C., Tara Oceans: towards global ocean ecosystems biology. Nat. Rev. Microbiol. 18, 428–445 (2020). - PubMed
-
- Nayfach S., Roux S., Seshadri R., Udwary D., Varghese N., Schulz F., Wu D., Paez-Espino D., Chen I.-M., Huntemann M., Palaniappan K., Ladau J., Mukherjee S., Reddy T. B. K., Nielsen T., Kirton E., Faria J. P., Edirisinghe J. N., Henry C. S., Jungbluth S. P., Chivian D., Dehal P., Wood-Charlson E. M., Arkin A. P., Tringe S. G., Visel A., IMG/M Data Consortium, Woyke T., Mouncey N. J., Ivanova N. N., Kyrpides N. C., Eloe-Fadrosh E. A., A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 39, 499–509 (2021). - PMC - PubMed
-
- Yatsunenko T., Rey F. E., Manary M. J., Trehan I., Dominguez-Bello M. G., Contreras M., Magris M., Hidalgo G., Baldassano R. N., Anokhin A. P., Heath A. C., Warner B., Reeder J., Kuczynski J., Caporaso J. G., Lozupone C. A., Lauber C., Clemente J. C., Knights D., Knight R., Gordon J. I., Human gut microbiome viewed across age and geography. Nature. 486, 222–227 (2012). - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources