

This is a preprint.
ProteinNPT: Improving Protein Property Prediction and Design with Non-Parametric Transformers
- PMID: 38106034
- PMCID: PMC10723423
- DOI: 10.1101/2023.12.06.570473
ProteinNPT: Improving Protein Property Prediction and Design with Non-Parametric Transformers
Abstract
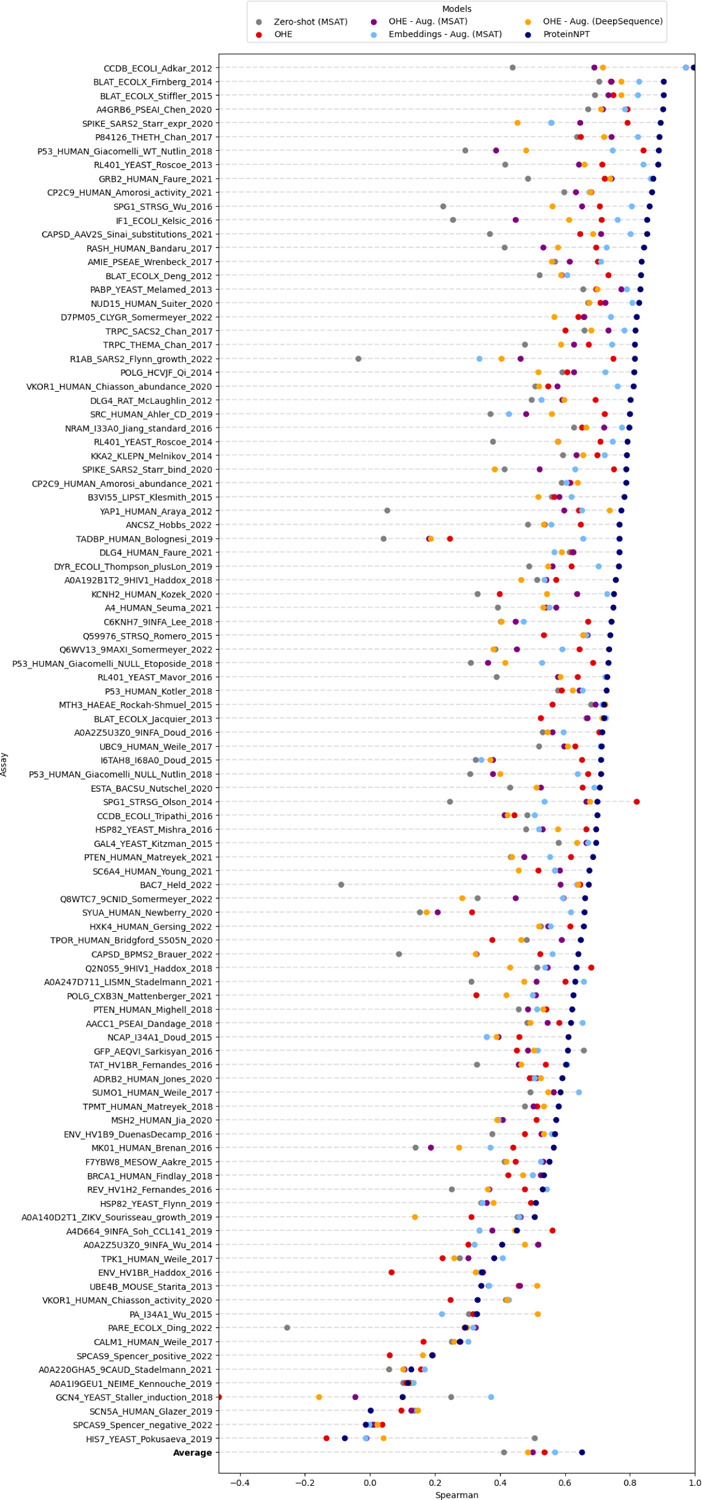
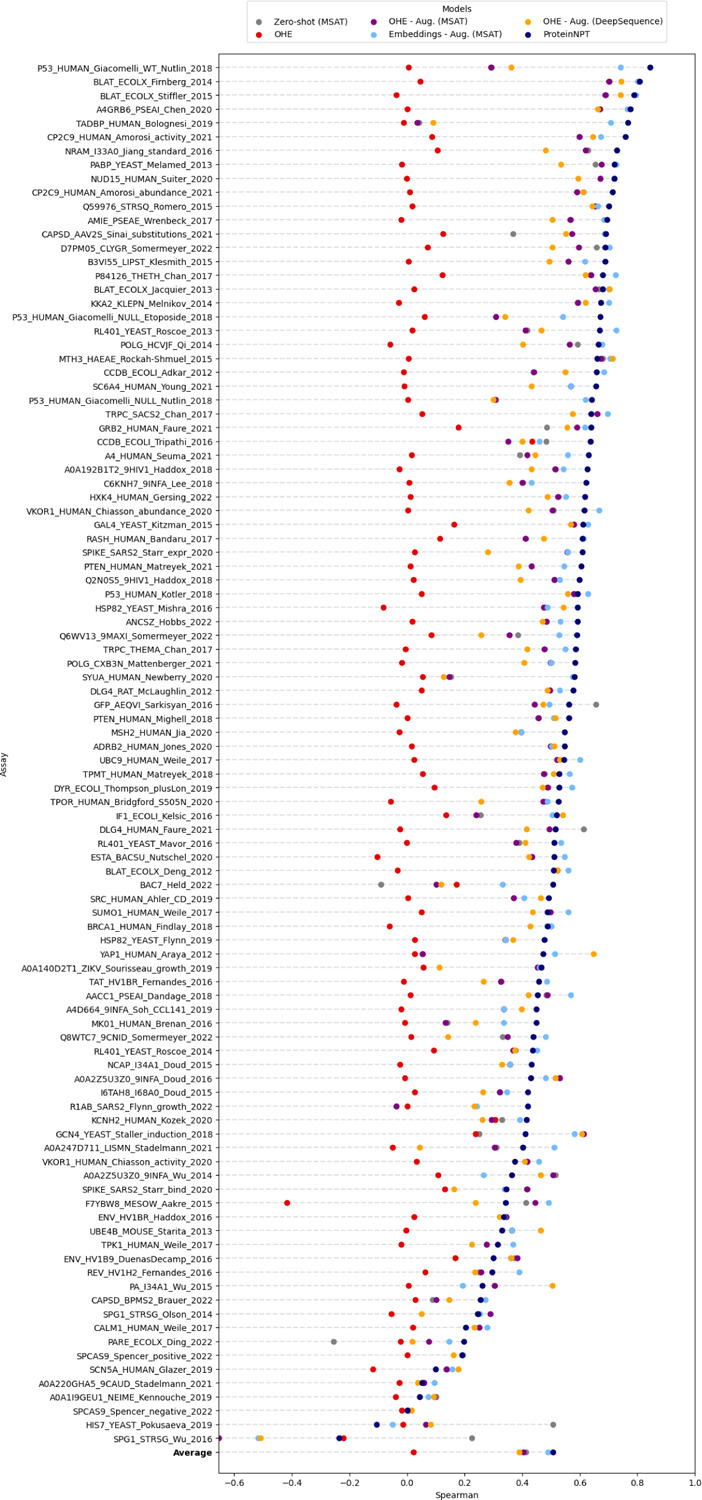
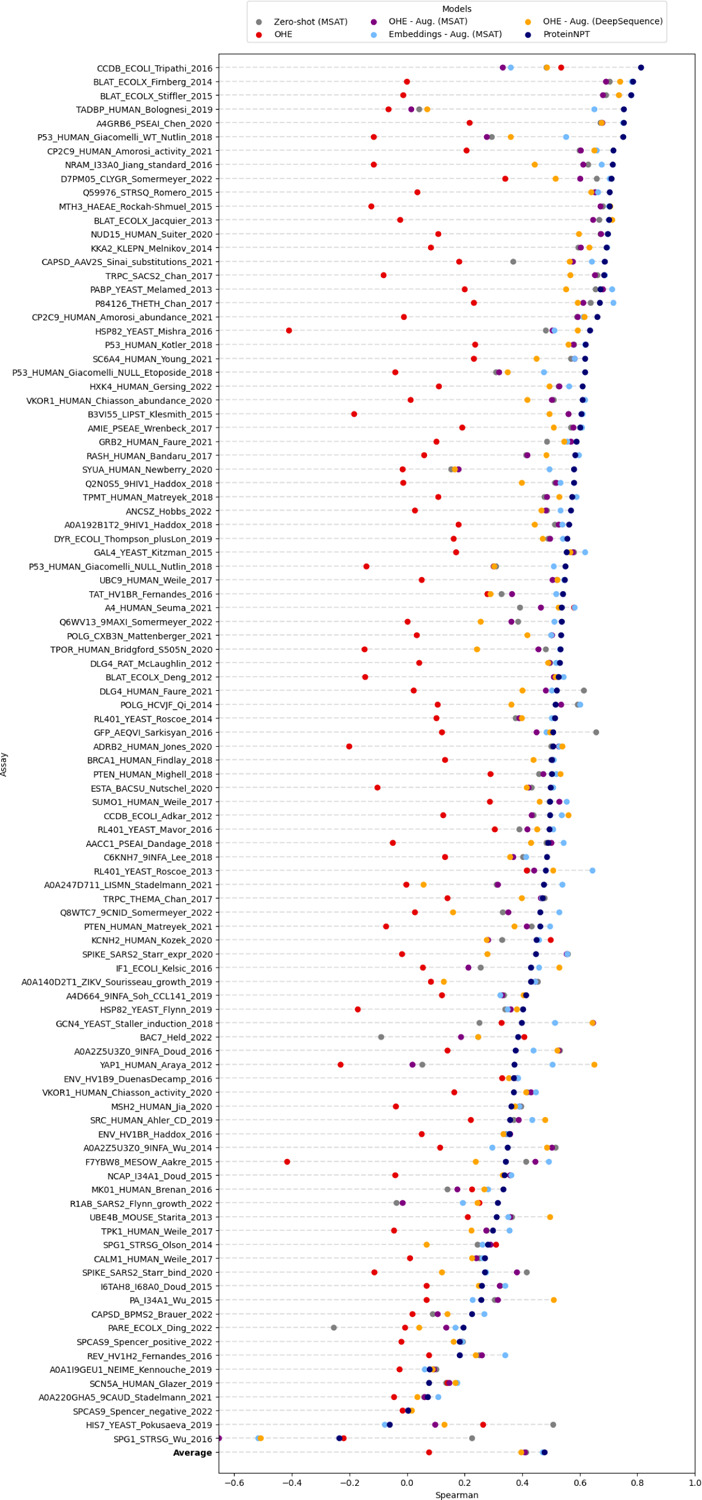
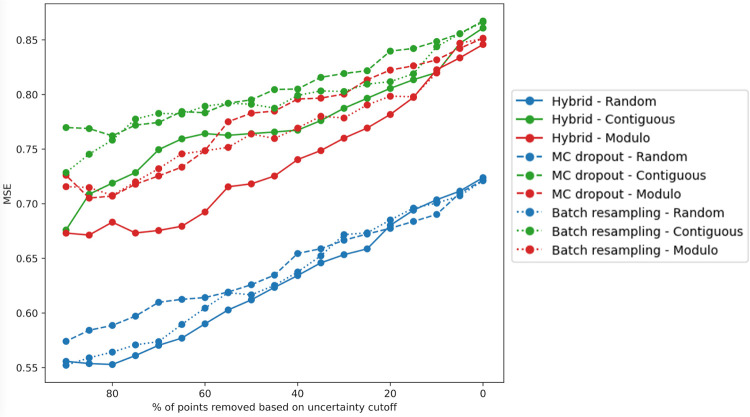
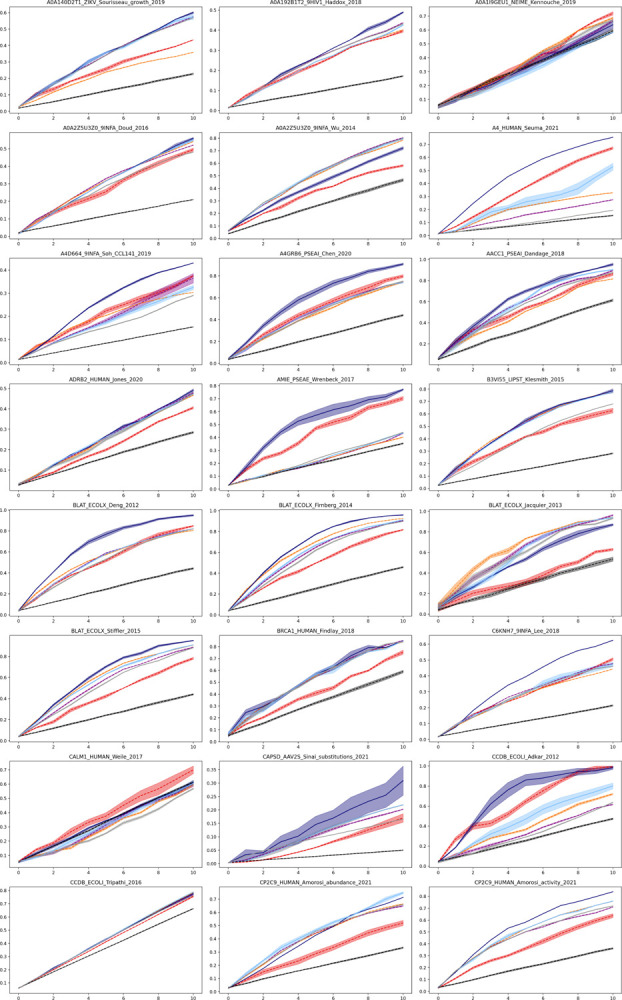
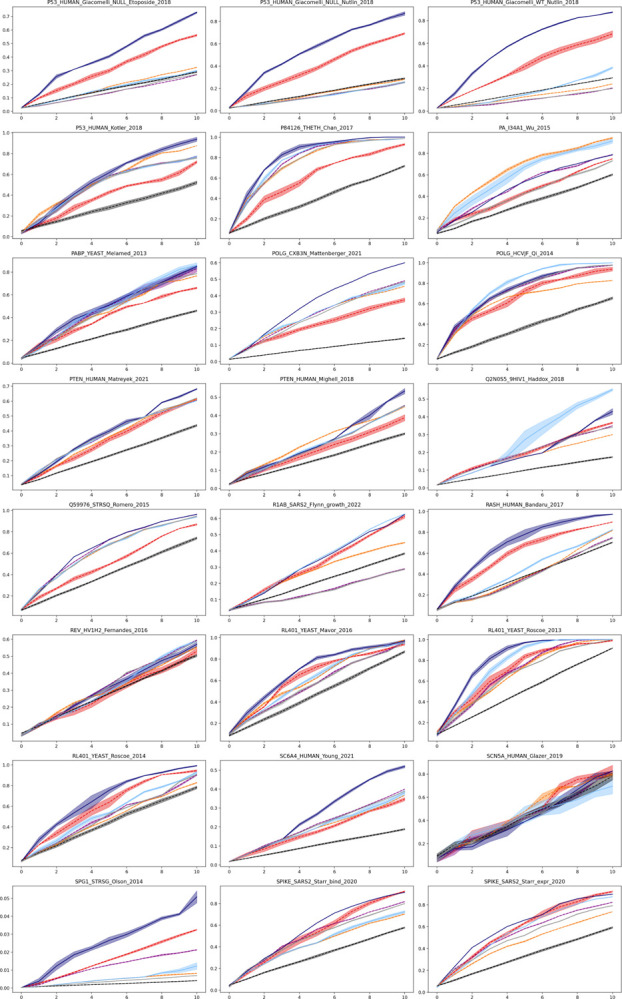
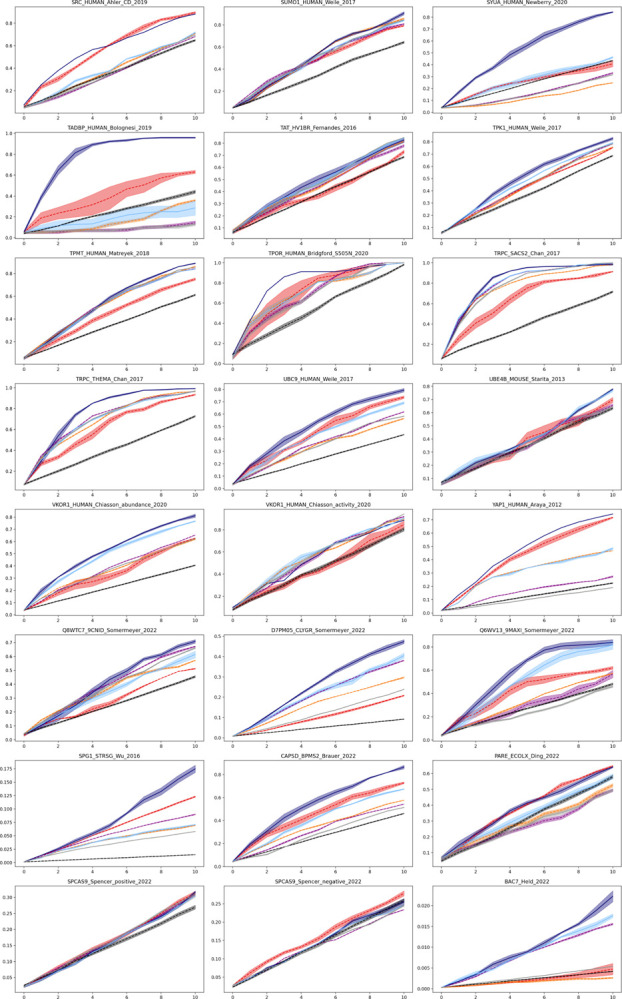
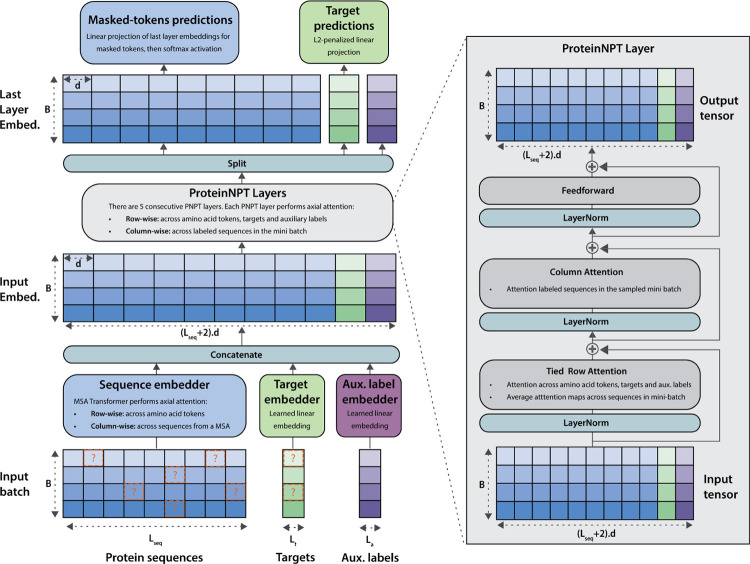
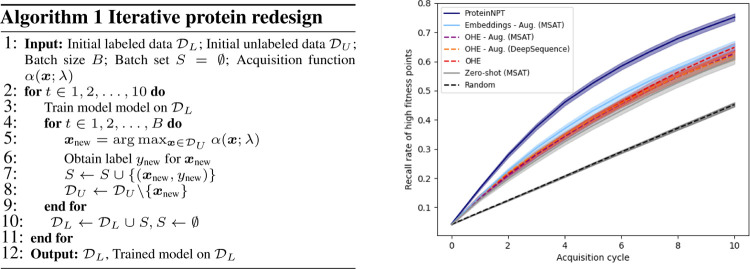
Protein design holds immense potential for optimizing naturally occurring proteins, with broad applications in drug discovery, material design, and sustainability. However, computational methods for protein engineering are confronted with significant challenges, such as an expansive design space, sparse functional regions, and a scarcity of available labels. These issues are further exacerbated in practice by the fact most real-life design scenarios necessitate the simultaneous optimization of multiple properties. In this work, we introduce ProteinNPT, a non-parametric transformer variant tailored to protein sequences and particularly suited to label-scarce and multi-task learning settings. We first focus on the supervised fitness prediction setting and develop several cross-validation schemes which support robust performance assessment. We subsequently reimplement prior top-performing baselines, introduce several extensions of these baselines by integrating diverse branches of the protein engineering literature, and demonstrate that ProteinNPT consistently outperforms all of them across a diverse set of protein property prediction tasks. Finally, we demonstrate the value of our approach for iterative protein design across extensive in silico Bayesian optimization and conditional sampling experiments.
Figures






References
-
- Arnold Frances H.. Directed Evolution: Bringing New Chemistry to Life. Angewandte Chemie International Edition, 57(16):4143–4148, 2018. ISSN 1521–3773. doi: 10.1002/anie.201708408. URL https://onlinelibrary.wiley.com/doi/abs/10.1002/anie.201708408._eprint: https://onlinelibrary.wiley.com/doi/pdf/10.1002/anie.201708408. - DOI - DOI - DOI - PMC - PubMed
-
- Huang Po-Ssu, Boyken Scott E., and Baker David. The coming of age of de novo protein design. Nature, 537(7620):320–327, September 2016. ISSN 1476–4687. doi: 10.1038/nature19946. URL https://www.nature.com/articles/nature19946. Number: 7620 Publisher: Nature Publishing Group. - DOI - PubMed
-
- Romero Philip A., Krause Andreas, and Arnold Frances H.. Navigating the protein fitness landscape with Gaussian processes. Proceedings of the National Academy of Sciences, 110(3):E193–E201, January 2013. doi: 10.1073/pnas.1215251110. URL https://www.pnas.org/doi/10.1073/pnas.1215251110. Publisher: Proceedings of the National Academy of Sciences. - DOI - DOI - PMC - PubMed
-
- Biswas Surojit, Khimulya Grigory, Alley Ethan C., Esvelt Kevin M., and Church George M.. Low-N protein engineering with data-efficient deep learning. Nature Methods, 18(4):389–396, April 2021a. ISSN 1548–7091, 1548–7105. doi: 10.1038/s41592-021-01100-y. URL http://www.nature.com/articles/s41592-021-01100-y. - DOI - PubMed
-
- Dougherty Michael J and Arnold Frances H. Directed evolution: new parts and optimized function. Current Opinion in Biotechnology, 20(4):486–491, August 2009. ISSN 0958–1669. doi: 10.1016/j.copbio.2009.08.005. URL https://www.sciencedirect.com/science/article/pii/S0958166909000986. - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources