

This is a preprint.
Systematic benchmarking of imaging spatial transcriptomics platforms in FFPE tissues
- PMID: 38106230
- PMCID: PMC10723440
- DOI: 10.1101/2023.12.07.570603
Systematic benchmarking of imaging spatial transcriptomics platforms in FFPE tissues
Abstract
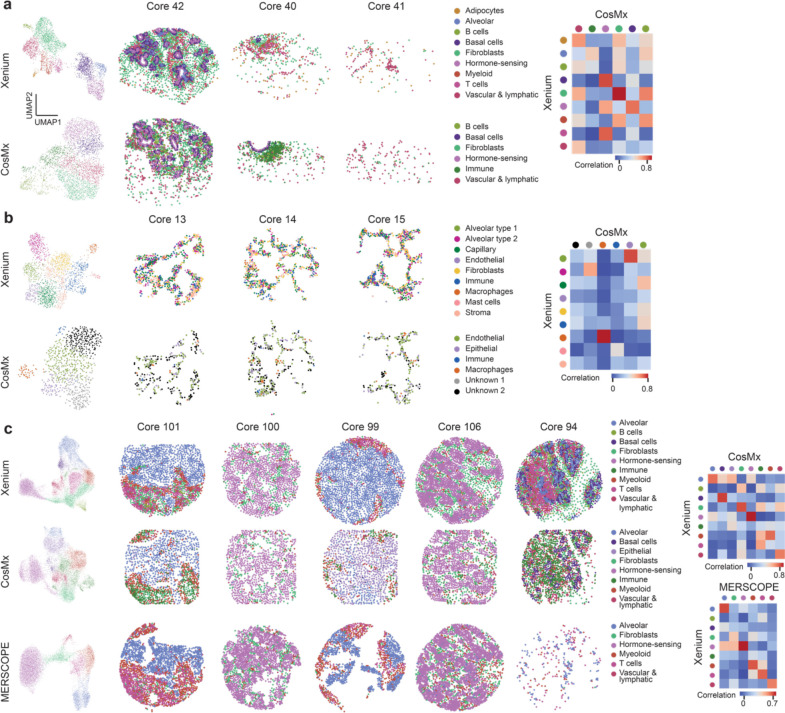
Emerging imaging spatial transcriptomics (iST) platforms and coupled analytical methods can recover cell-to-cell interactions, groups of spatially covarying genes, and gene signatures associated with pathological features, and are thus particularly well-suited for applications in formalin fixed paraffin embedded (FFPE) tissues. Here, we benchmarked the performance of three commercial iST platforms on serial sections from tissue microarrays (TMAs) containing 23 tumor and normal tissue types for both relative technical and biological performance. On matched genes, we found that 10x Xenium shows higher transcript counts per gene without sacrificing specificity, but that all three platforms concord to orthogonal RNA-seq datasets and can perform spatially resolved cell typing, albeit with different false discovery rates, cell segmentation error frequencies, and with varying degrees of sub-clustering for downstream biological analyses. Taken together, our analyses provide a comprehensive benchmark to guide the choice of iST method as researchers design studies with precious samples in this rapidly evolving field.
Conflict of interest statement
Declaration of interests All authors declare that they have no conflicts of interest.
Figures
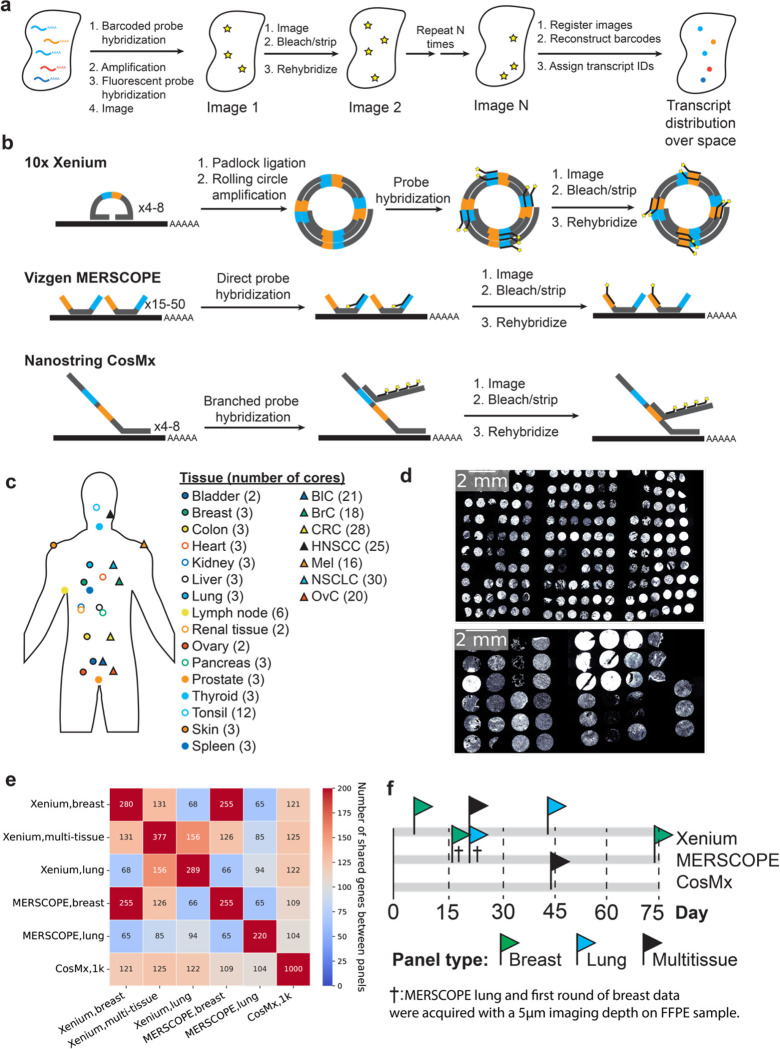
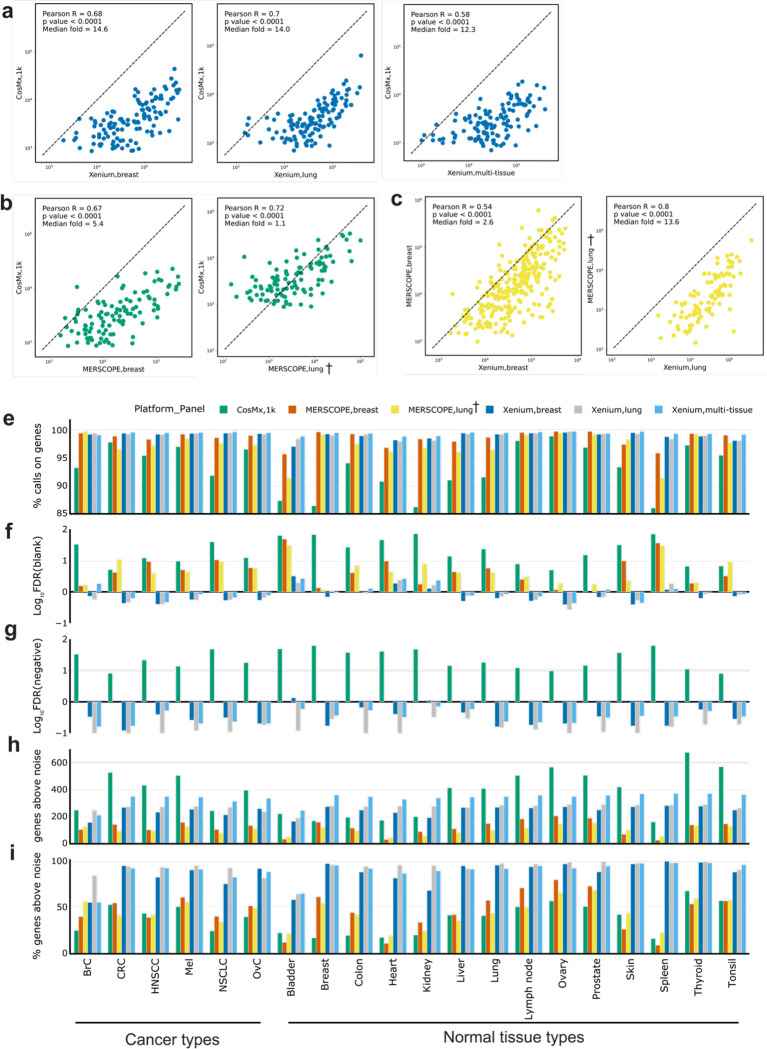
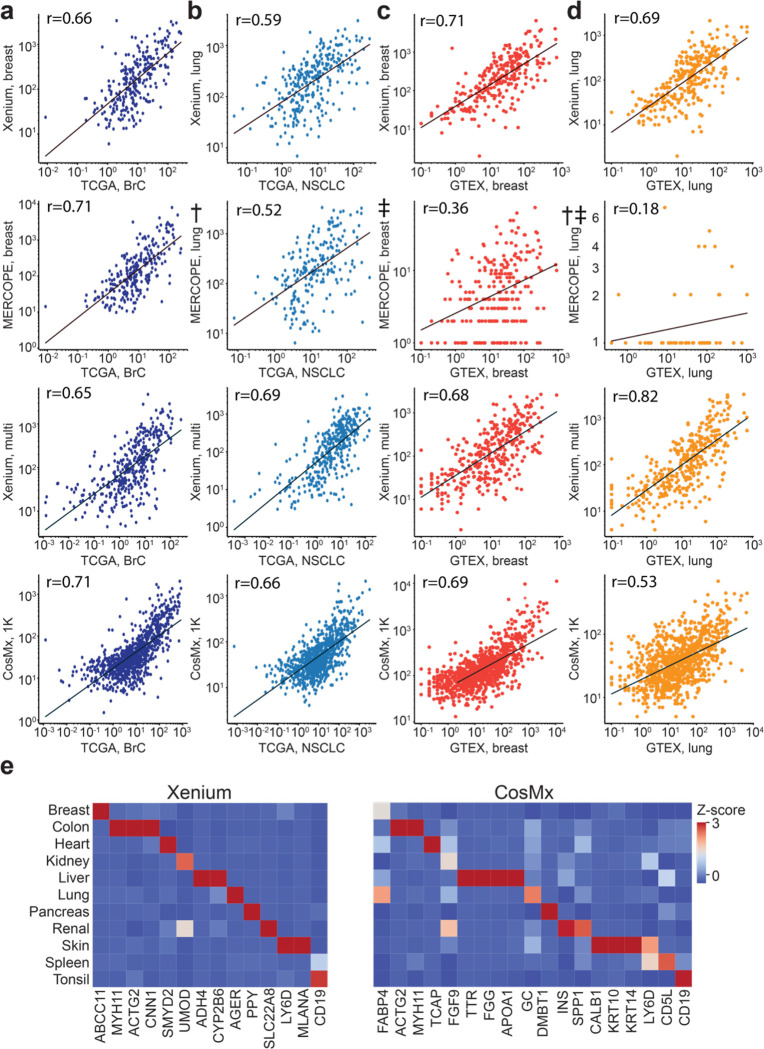
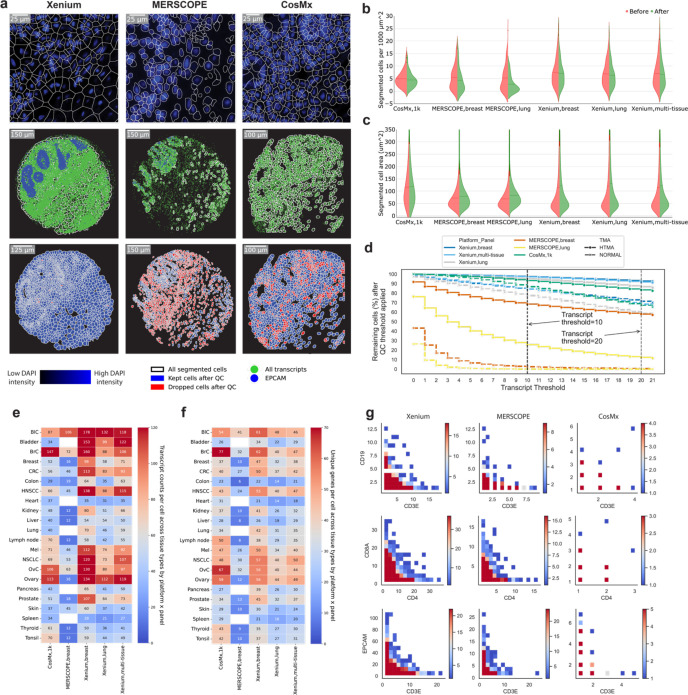
References
-
- Chen W.T., Lu A. Croissants K. et al. Spatial Transcriptomics and In Situ Sequencing to Study Alzheimer’s Disease. Cell. 182 (4), 976–991 (2020). - PubMed
-
- Lein E, Borm LE, Linnarsson S. The promise of spatial transcriptomics for neuroscience in the era of molecular cell typing. Science. 2017. Oct 6;358(6359):64–69. - PubMed
-
- Lewis SM, Asselin-Labat ML, Nguyen Q, et al. Spatial omics and multiplexed imaging to explore cancer biology. Nat Methods. 2021. Sep;18(9):997–1012. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources