

This is a preprint.
SOS1 inhibition enhances the efficacy of and delays resistance to G12C inhibitors in lung adenocarcinoma
- PMID: 38106234
- PMCID: PMC10723384
- DOI: 10.1101/2023.12.07.570642
SOS1 inhibition enhances the efficacy of and delays resistance to G12C inhibitors in lung adenocarcinoma
Update in
-
SOS1 Inhibition Enhances the Efficacy of KRASG12C Inhibitors and Delays Resistance in Lung Adenocarcinoma.Cancer Res. 2025 Jan 2;85(1):118-133. doi: 10.1158/0008-5472.CAN-23-3256. Cancer Res. 2025. PMID: 39437166 Free PMC article.
Abstract
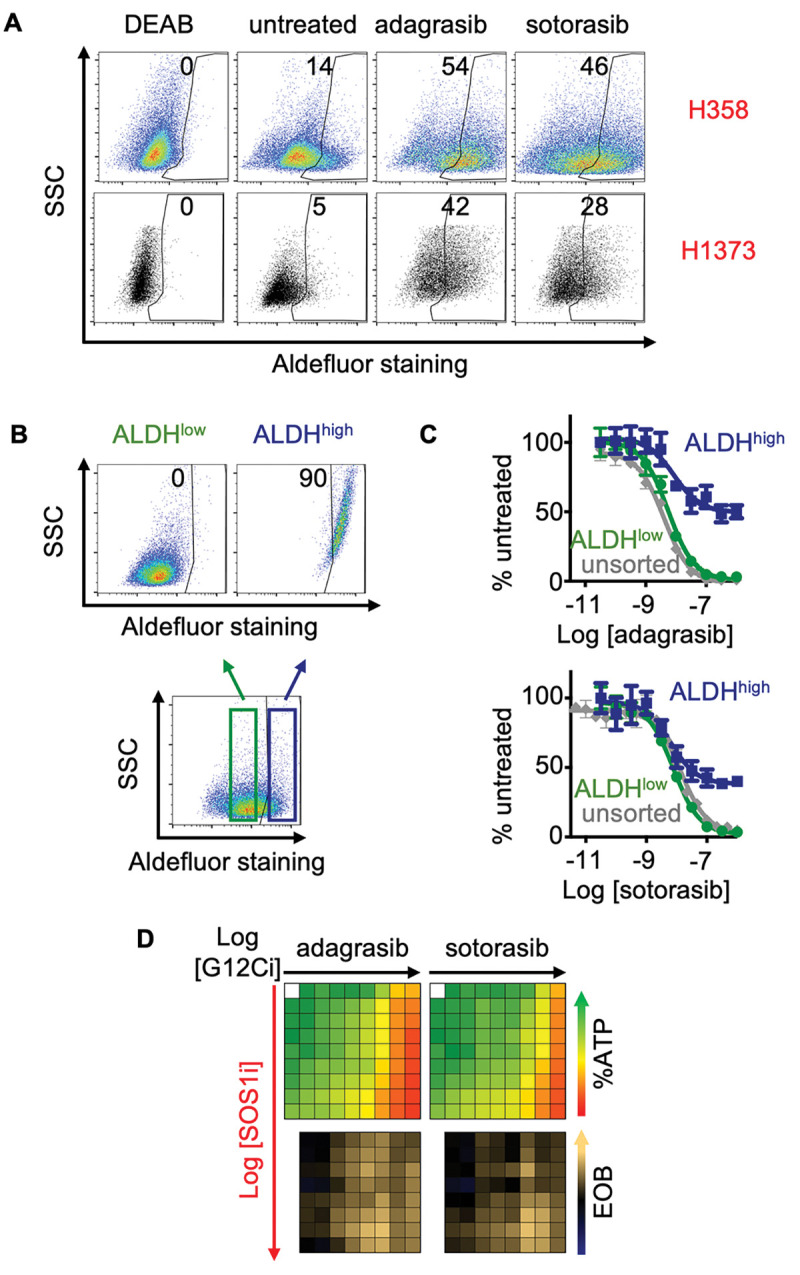
Clinical effectiveness of KRAS G12C inhibitors (G12Cis) is limited both by intrinsic and acquired resistance, necessitating the development of combination approaches. We found that targeting proximal receptor tyrosine kinase (RTK) signaling using the SOS1 inhibitor (SOS1i) BI-3406 both enhanced the potency of and delayed resistance to G12Ci treatment, but the extent of SOS1i effectiveness was modulated by both SOS2 expression and the specific mutational landscape. SOS1i enhanced the efficacy of G12Ci and limited rebound RTK/ERK signaling to overcome intrinsic/adaptive resistance, but this effect was modulated by SOS2 protein levels. Survival of drug-tolerant persister (DTP) cells within the heterogeneous tumor population and/or acquired mutations that reactivate RTK/RAS signaling can lead to outgrowth of tumor initiating cells (TICs) that drive therapeutic resistance. G12Ci drug tolerant persister cells showed a 2-3-fold enrichment of TICs, suggesting that these could be a sanctuary population of G12Ci resistant cells. SOS1i re-sensitized DTPs to G12Ci and inhibited G12C-induced TIC enrichment. Co-mutation of the tumor suppressor KEAP1 limits the clinical effectiveness of G12Cis, and KEAP1 and STK11 deletion increased TIC frequency and accelerated the development of acquired resistance to G12Ci in situ. SOS1i both delayed acquired G12Ci resistance and limited the total number of resistant colonies regardless of KEAP1 and STK11 mutational status. These data suggest that SOS1i could be an effective strategy to both enhance G12Ci efficacy and prevent G12Ci resistance regardless of co-mutations.
Figures






References
-
- Gridelli C, Rossi A, Carbone DP, Guarize J, Karachaliou N, Mok T, et al. Non-small-cell lung cancer. Nat Rev Dis Primers 2015;1:15009. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous