

Targeting RACK1 to alleviate TDP-43 and FUS proteinopathy-mediated suppression of protein translation and neurodegeneration
- PMID: 38111057
- PMCID: PMC10726565
- DOI: 10.1186/s40478-023-01705-8
Targeting RACK1 to alleviate TDP-43 and FUS proteinopathy-mediated suppression of protein translation and neurodegeneration
Abstract
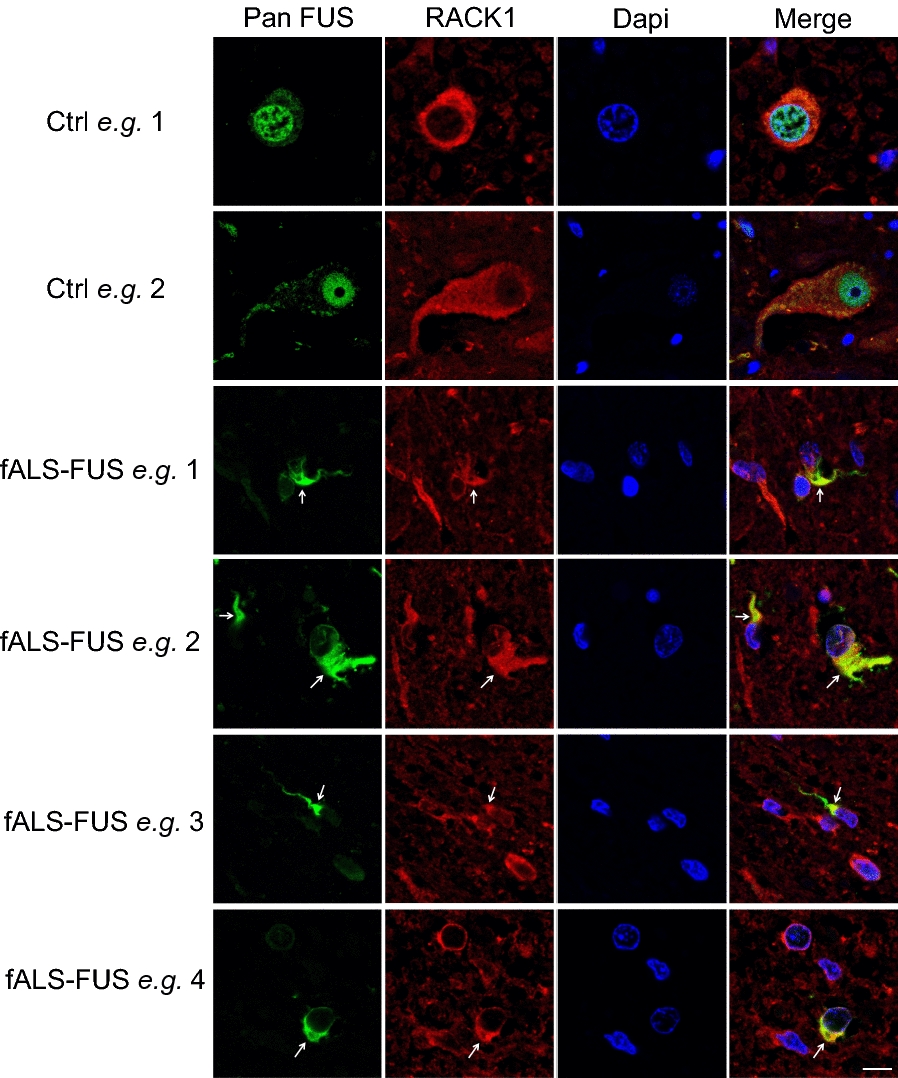
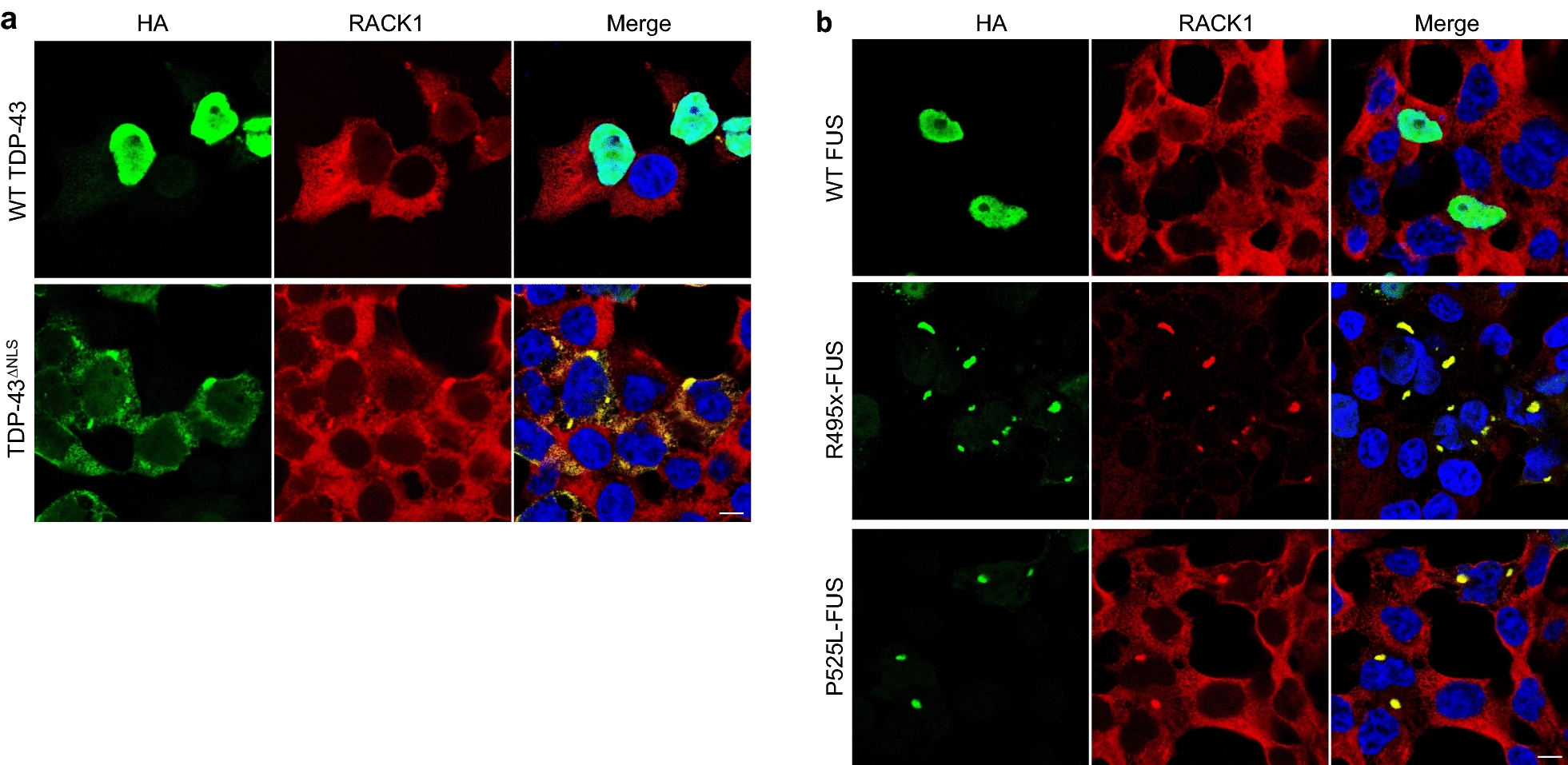
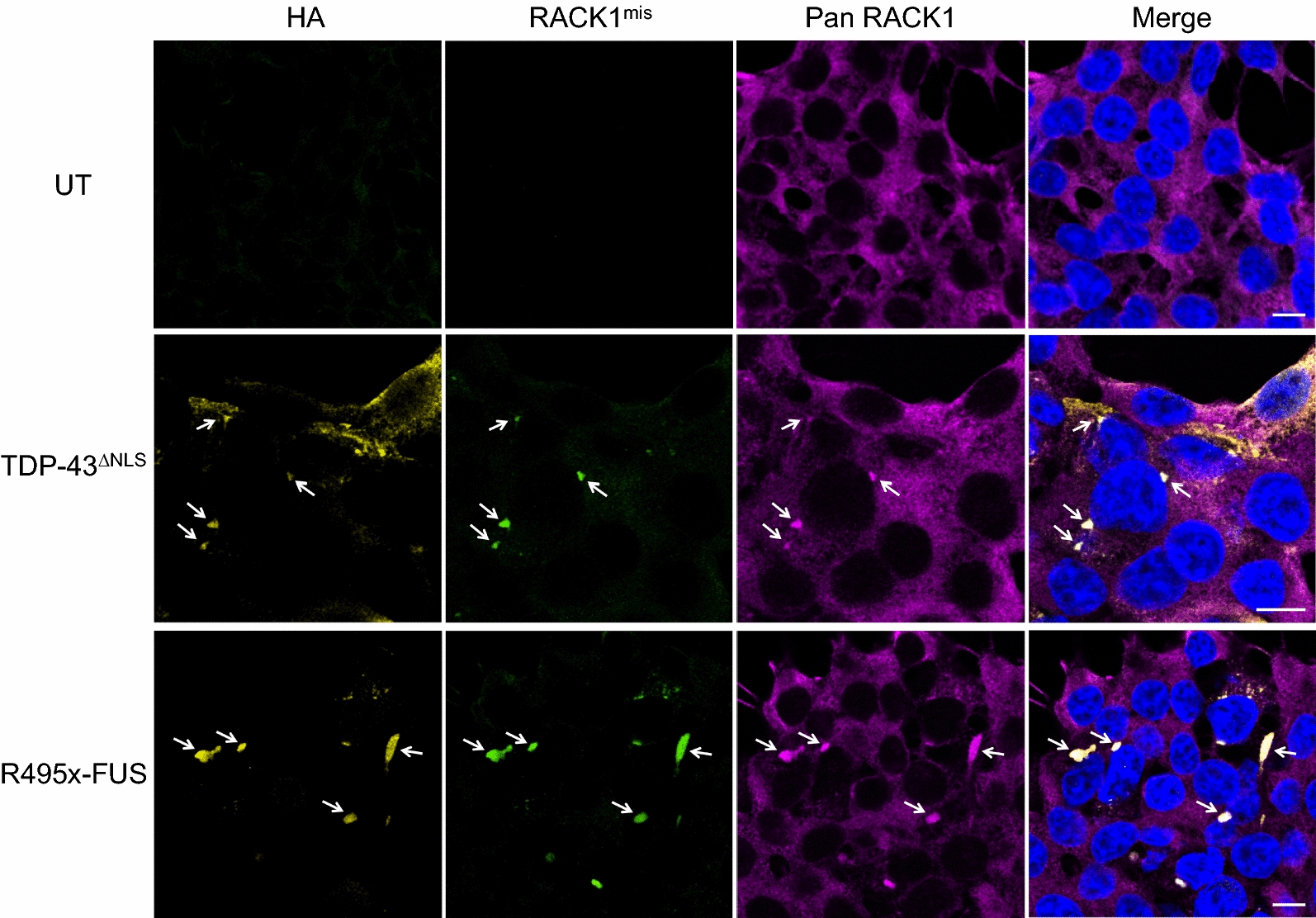
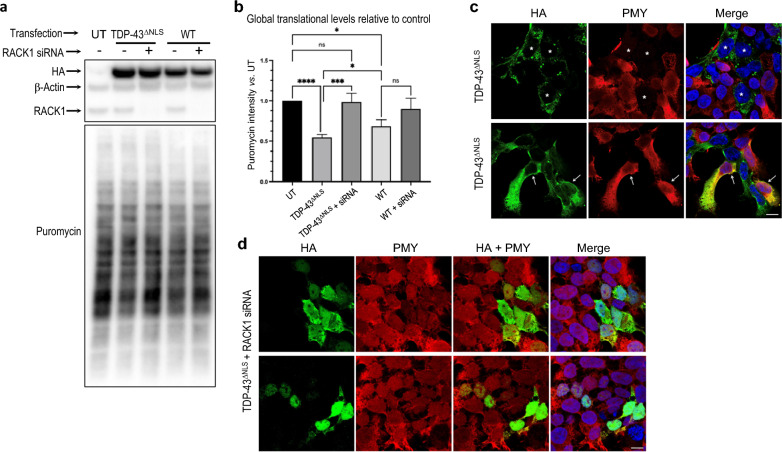
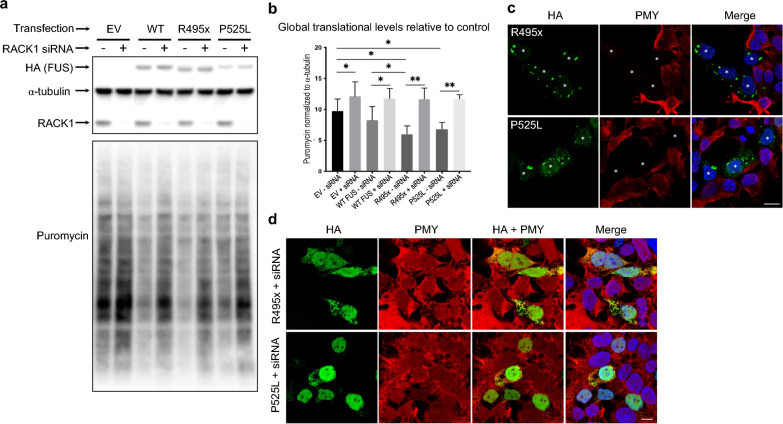
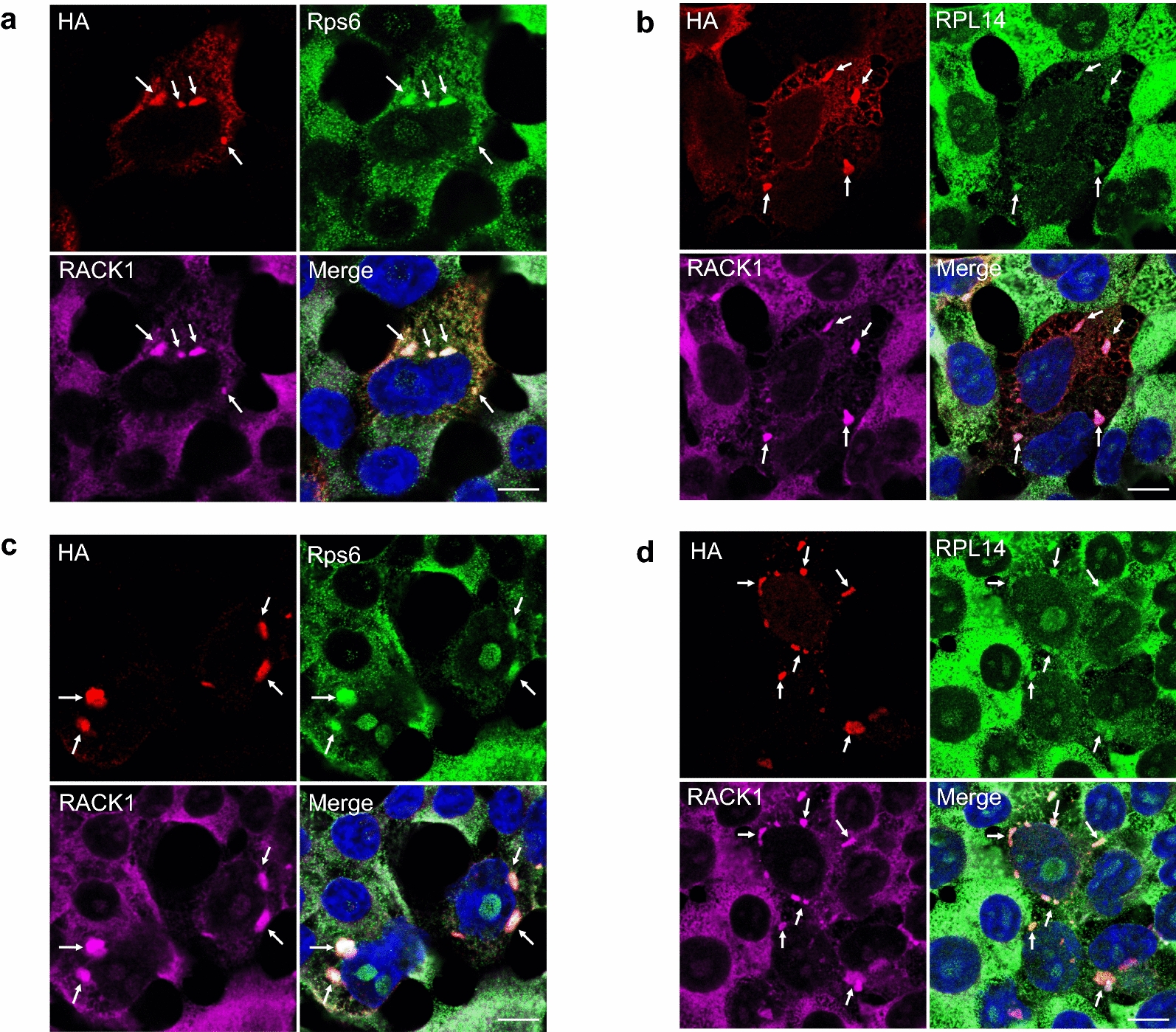
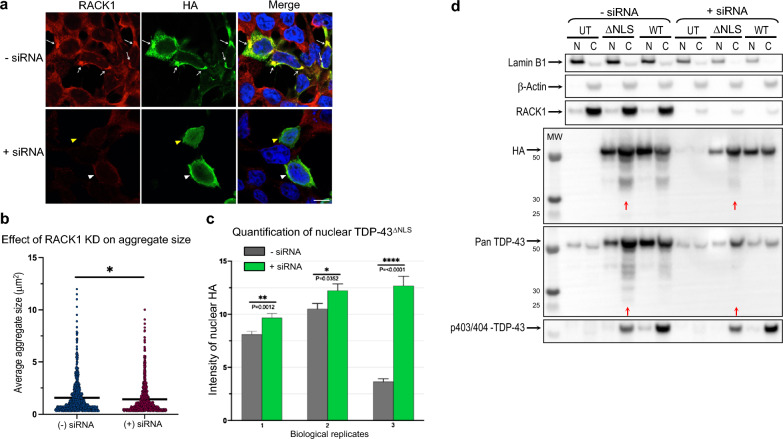
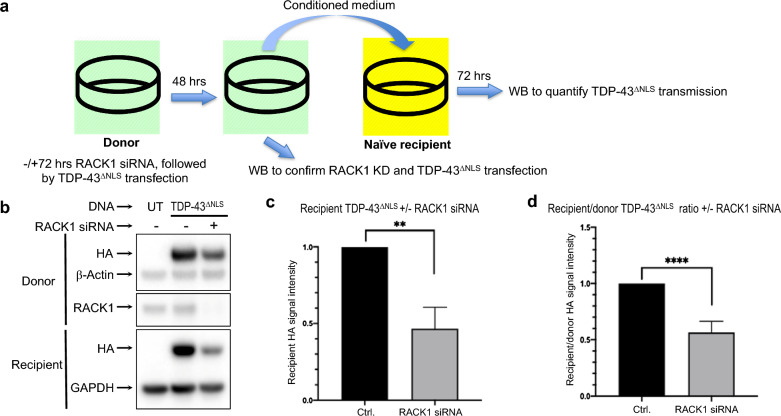
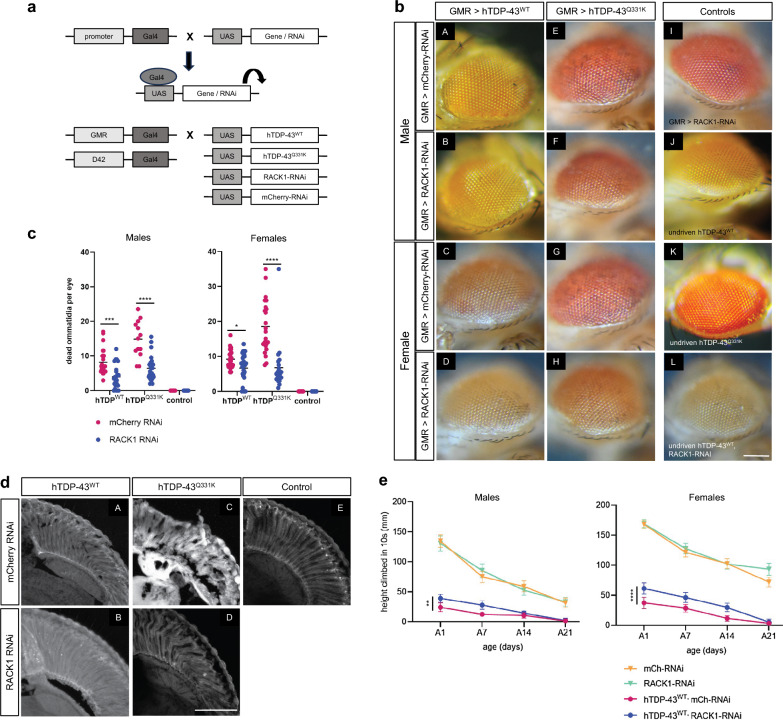
TAR DNA-binding protein 43 (TDP-43) and Fused in Sarcoma/Translocated in Sarcoma (FUS) are ribonucleoproteins associated with pathogenesis of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). Under physiological conditions, TDP-43 and FUS are predominantly localized in the nucleus, where they participate in transcriptional regulation, RNA splicing and metabolism. In disease, however, they are typically mislocalized to the cytoplasm where they form aggregated inclusions. A number of shared cellular pathways have been identified that contribute to TDP-43 and FUS toxicity in neurodegeneration. In the present study, we report a novel pathogenic mechanism shared by these two proteins. We found that pathological FUS co-aggregates with a ribosomal protein, the Receptor for Activated C-Kinase 1 (RACK1), in the cytoplasm of spinal cord motor neurons of ALS, as previously reported for pathological TDP-43. In HEK293T cells transiently transfected with TDP-43 or FUS mutant lacking a functional nuclear localization signal (NLS; TDP-43ΔNLS and FUSΔNLS), cytoplasmic TDP-43 and FUS induced co-aggregation with endogenous RACK1. These co-aggregates sequestered the translational machinery through interaction with the polyribosome, accompanied by a significant reduction of global protein translation. RACK1 knockdown decreased cytoplasmic aggregation of TDP-43ΔNLS or FUSΔNLS and alleviated associated global translational suppression. Surprisingly, RACK1 knockdown also led to partial nuclear localization of TDP-43ΔNLS and FUSΔNLS in some transfected cells, despite the absence of NLS. In vivo, RACK1 knockdown alleviated retinal neuronal degeneration in transgenic Drosophila melanogaster expressing hTDP-43WT or hTDP-43Q331K and improved motor function of hTDP-43WT flies, with no observed adverse effects on neuronal health in control knockdown flies. In conclusion, our results revealed a novel shared mechanism of pathogenesis for misfolded aggregates of TDP-43 and FUS mediated by interference with protein translation in a RACK1-dependent manner. We provide proof-of-concept evidence for targeting RACK1 as a potential therapeutic approach for TDP-43 or FUS proteinopathy associated with ALS and FTLD.
© 2023. The Author(s).
Conflict of interest statement
N.R.C., J.M.K., and B.Z. are current employees of ProMIS Neurosciences. S.S.P. and S.C.C.H. have received consultation compensation from ProMIS Neurosciences. S.S.P. and B.Z. possess ProMIS stock options. N.R.C. and J.M.K. possess ProMIS shares and stock options. S.S.P. is the inventor on patent applications for Collective Coordinates computational modeling. N.R.C., S.S.P., J.M.K., and B.Z. are inventors on patent applications relating to conformation-specific epitopes in RACK1, antibodies thereto and methods related thereof.
Figures
References
-
- Afroz T, Hock EM, Ernst P, Foglieni C, Jambeau M, Gilhespy LAB, Laferriere F, Maniecka Z, Pluckthun A, Mittl P, et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat Commun. 2017;8:45. doi: 10.1038/s41467-017-00062-0. - DOI - PMC - PubMed
-
- Arai T, Hasegawa M, Nonoka T, Kametani F, Yamashita M, Hosokawa M, Niizato K, Tsuchiya K, Kobayashi Z, Ikeda K, et al. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology. 2010;30:170–181. doi: 10.1111/j.1440-1789.2009.01089.x. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
