

Nonsense Variant PRDM16-Q187X Causes Impaired Myocardial Development and TGF-β Signaling Resulting in Noncompaction Cardiomyopathy in Humans and Mice
- PMID: 38113297
- PMCID: PMC10752244
- DOI: 10.1161/CIRCHEARTFAILURE.122.010351
Nonsense Variant PRDM16-Q187X Causes Impaired Myocardial Development and TGF-β Signaling Resulting in Noncompaction Cardiomyopathy in Humans and Mice
Abstract
Background: PRDM16 plays a role in myocardial development through TGF-β (transforming growth factor-beta) signaling. Recent evidence suggests that loss of PRDM16 expression is associated with cardiomyopathy development in mice, although its role in human cardiomyopathy development is unclear. This study aims to determine the impact of PRDM16 loss-of-function variants on cardiomyopathy in humans.
Methods: Individuals with PRDM16 variants were identified and consented. Induced pluripotent stem cell-derived cardiomyocytes were generated from a proband hosting a Q187X nonsense variant as an in vitro model and underwent proliferative and transcriptional analyses. CRISPR (clustered regularly interspaced short palindromic repeats)-mediated knock-in mouse model hosting the Prdm16Q187X allele was generated and subjected to ECG, histological, and transcriptional analysis.
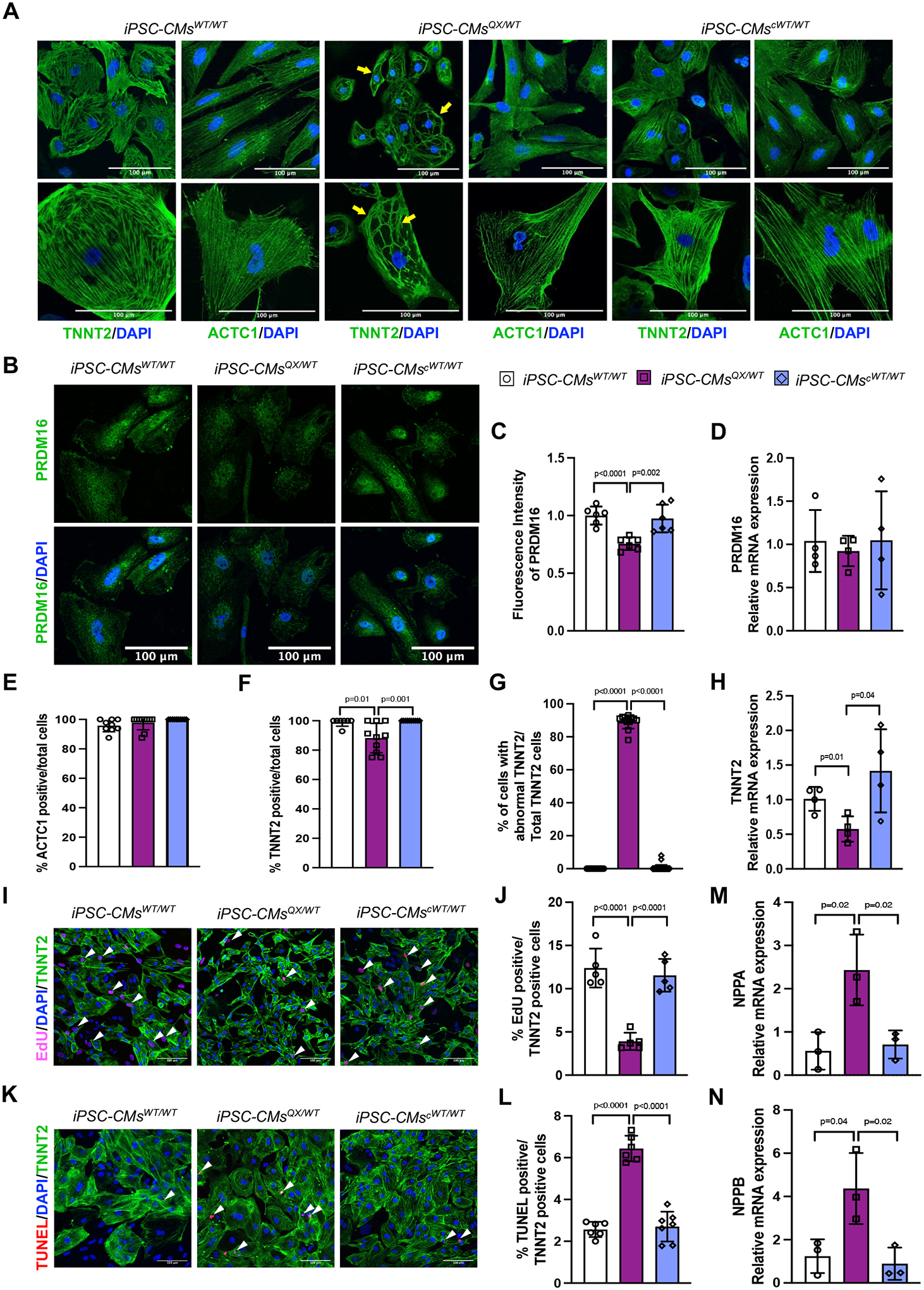
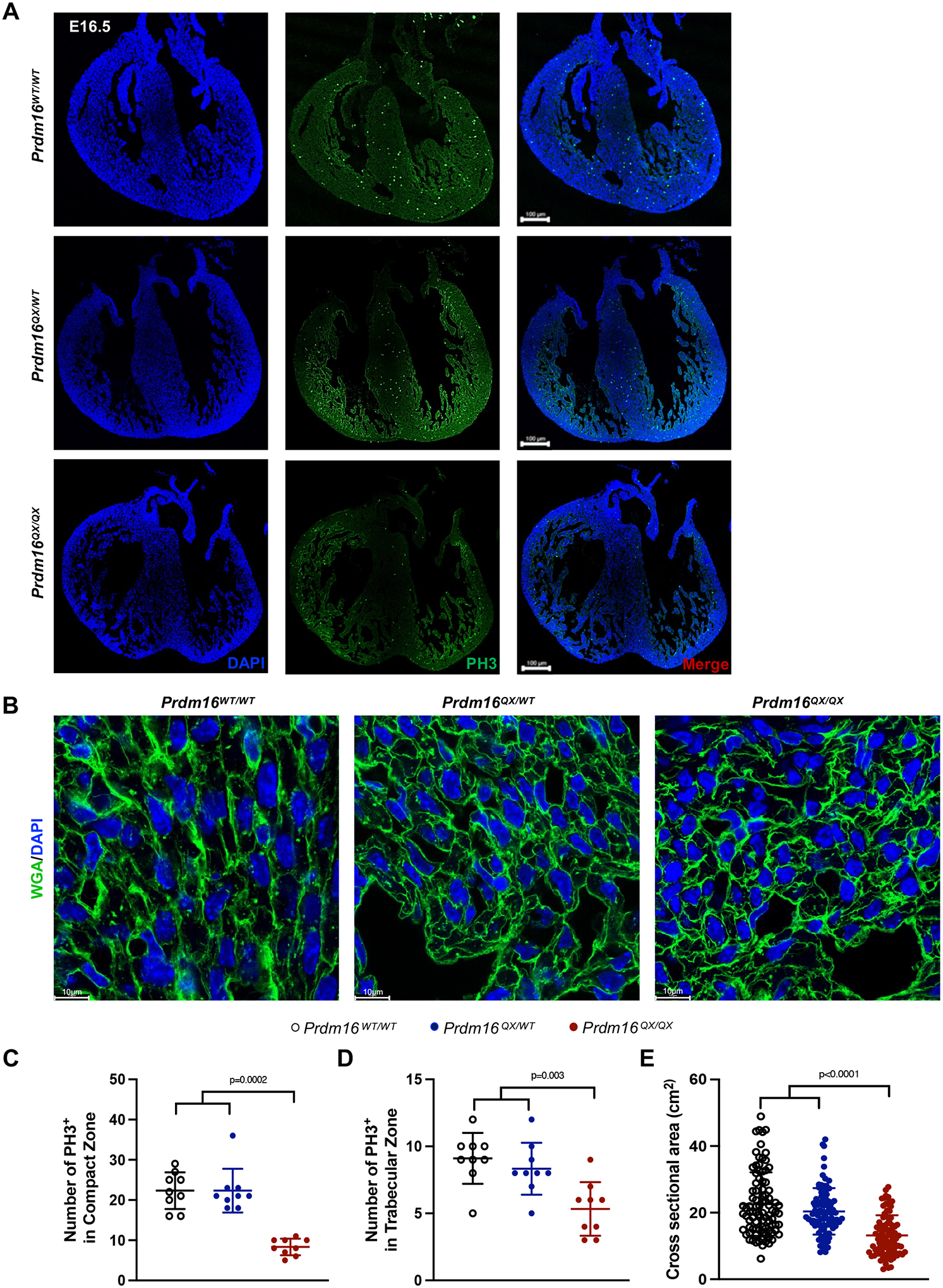
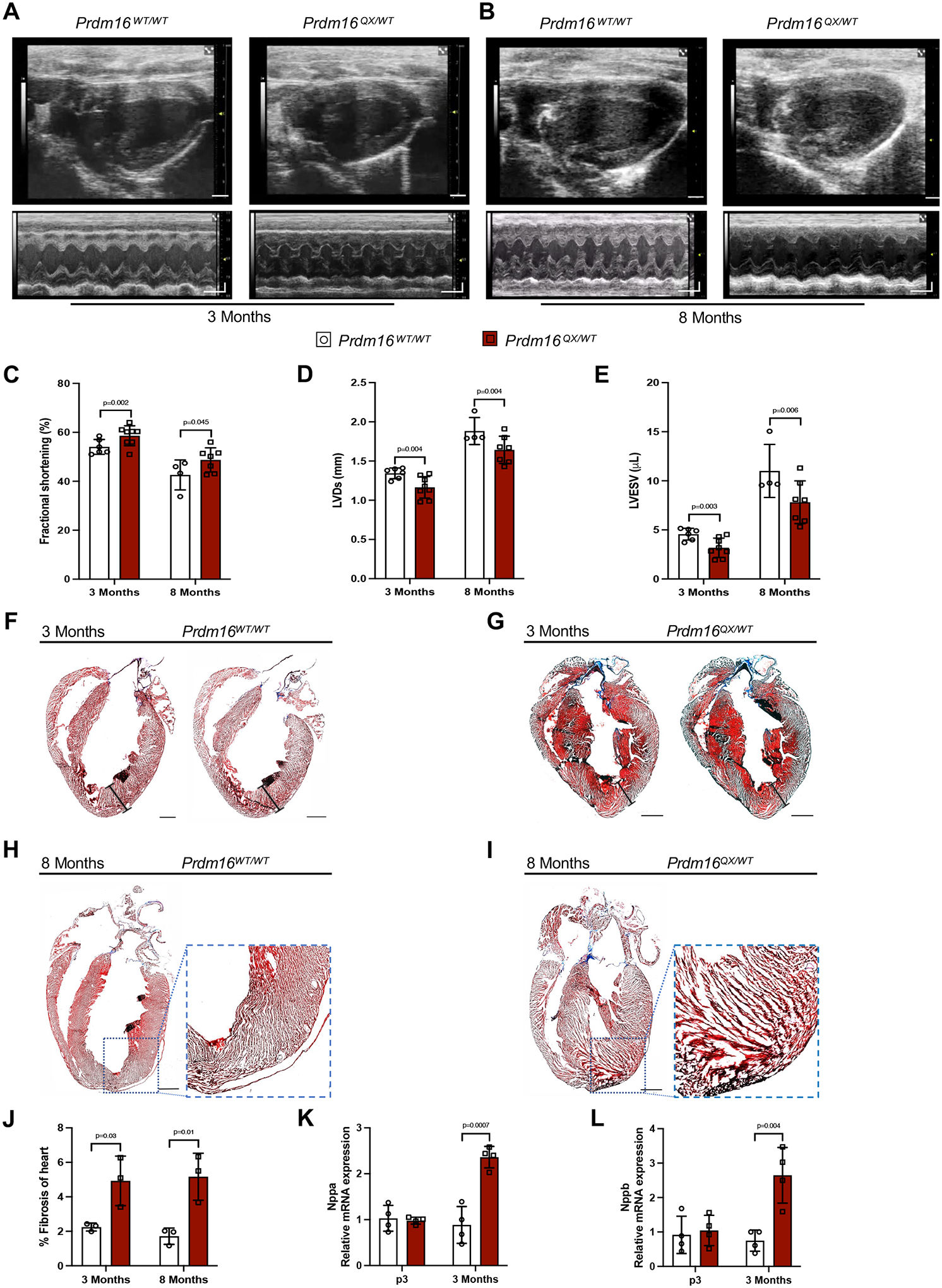
Results: We report 2 probands with loss-of-function PRDM16 variants and pediatric left ventricular noncompaction cardiomyopathy. One proband hosts a PRDM16-Q187X variant with left ventricular noncompaction cardiomyopathy and demonstrated infant-onset heart failure, which was selected for further study. Induced pluripotent stem cell-derived cardiomyocytes prepared from the PRDM16-Q187X proband demonstrated a statistically significant impairment in myocyte proliferation and increased apoptosis associated with transcriptional dysregulation of genes implicated in cardiac maturation, including TGF-β-associated transcripts. Homozygous Prdm16Q187X/Q187X mice demonstrated an underdeveloped compact myocardium and were embryonically lethal. Heterozygous Prdm16Q187X/WT mice demonstrated significantly smaller ventricular dimensions, heightened fibrosis, and age-dependent loss of TGF-β expression. Mechanistic studies were undertaken in H9c2 cardiomyoblasts to show that PRDM16 binds TGFB3 promoter and represses its transcription.
Conclusions: Novel loss-of-function PRDM16 variant impairs myocardial development resulting in noncompaction cardiomyopathy in humans and mice associated with altered TGF-β signaling.
Keywords: alleles; apoptosis; cardiomyopathies; induced pluripotent stem cells; mice; transcription factors.
Conflict of interest statement
Figures





References
-
- Towbin JA, Lorts A, and Jefferies JL, Left ventricular non-compaction cardiomyopathy. Lancet, 2015. 386(9995): p. 813–25. - PubMed
-
- Greutmann M, et al., Predictors of adverse outcome in adolescents and adults with isolated left ventricular noncompaction. Am J Cardiol, 2012. 109(2): p. 276–81. - PubMed
-
- Klaassen S, et al., Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation, 2008. 117(22): p. 2893–901. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- T32 HL139430/HL/NHLBI NIH HHS/United States
- R01 HL149870/HL/NHLBI NIH HHS/United States
- F32 HL144034/HL/NHLBI NIH HHS/United States
- K08 HL136839/HL/NHLBI NIH HHS/United States
- R01 HL160654/HL/NHLBI NIH HHS/United States
- R01 HL089598/HL/NHLBI NIH HHS/United States
- R01 HL147108/HL/NHLBI NIH HHS/United States
- P30 DK020579/DK/NIDDK NIH HHS/United States
- R01 HL166217/HL/NHLBI NIH HHS/United States
- R01 HL153350/HL/NHLBI NIH HHS/United States
- T35 DK103596/DK/NIDDK NIH HHS/United States
- R01 HL121700/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
