

High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis
- PMID: 38114474
- PMCID: PMC10730913
- DOI: 10.1038/s41467-023-43458-x
High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis
Abstract
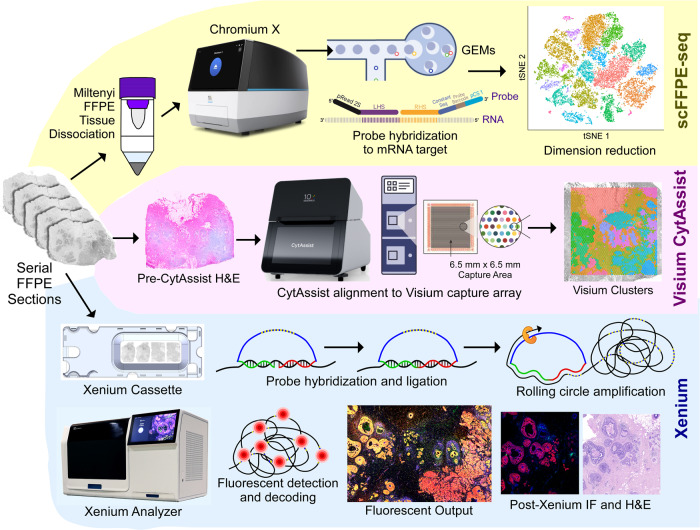
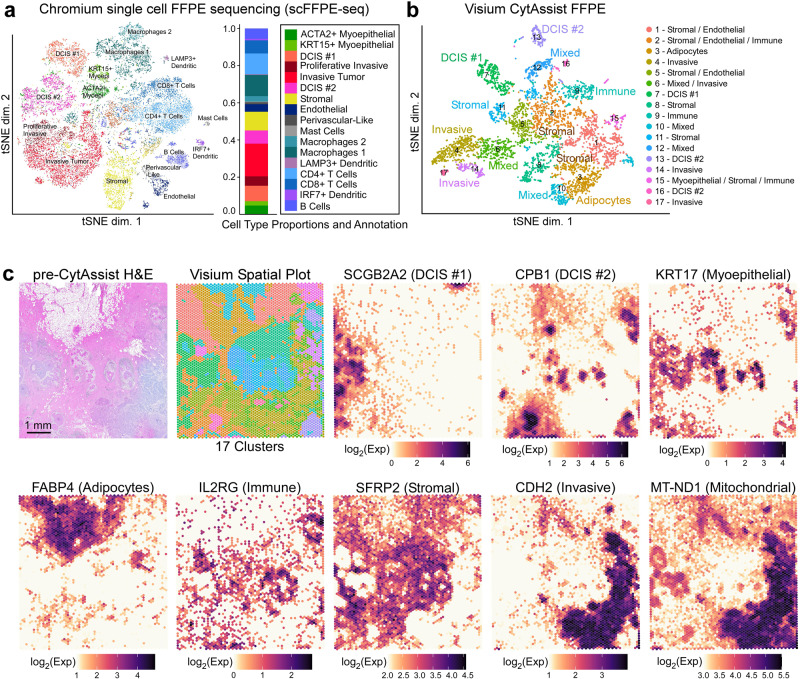
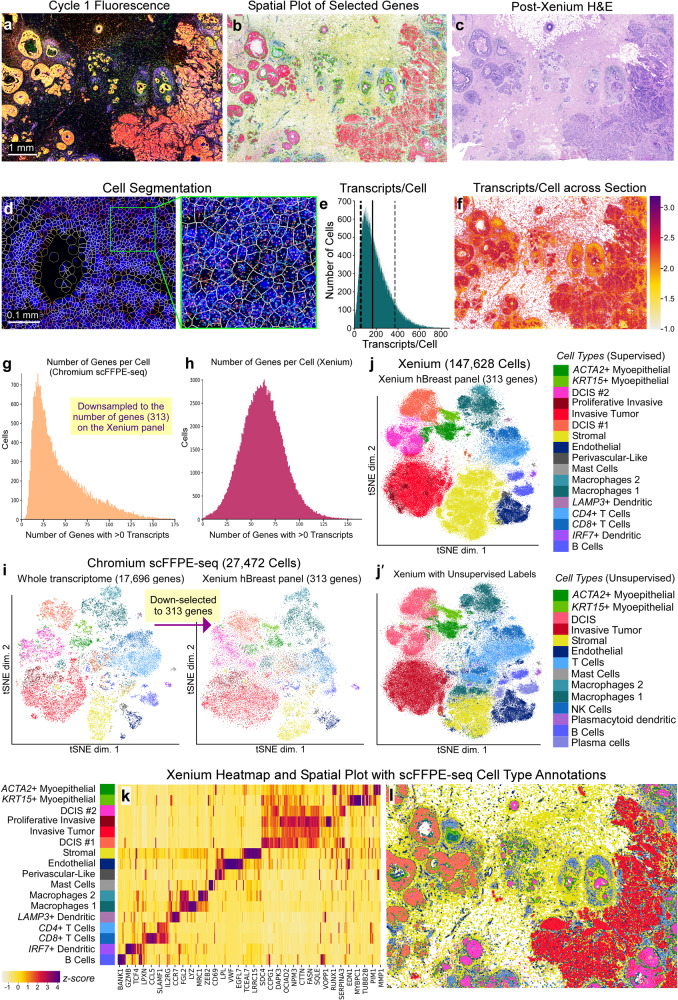
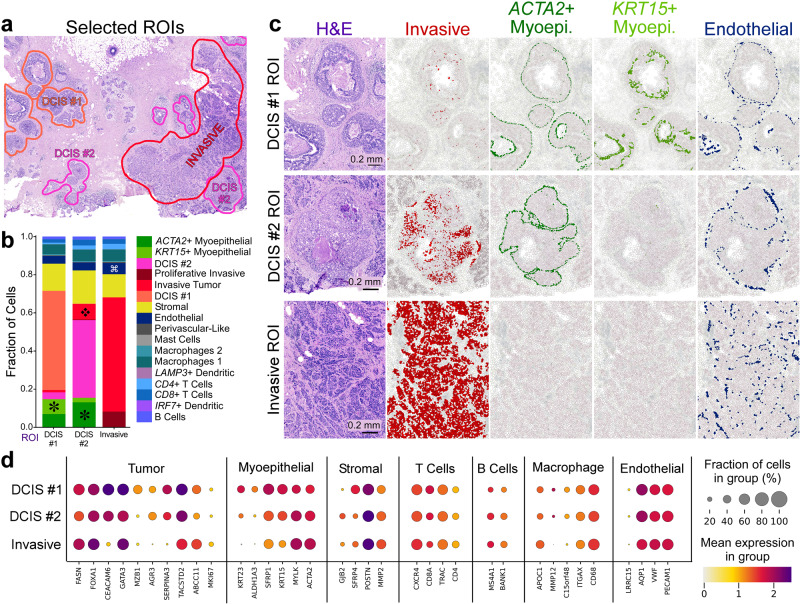
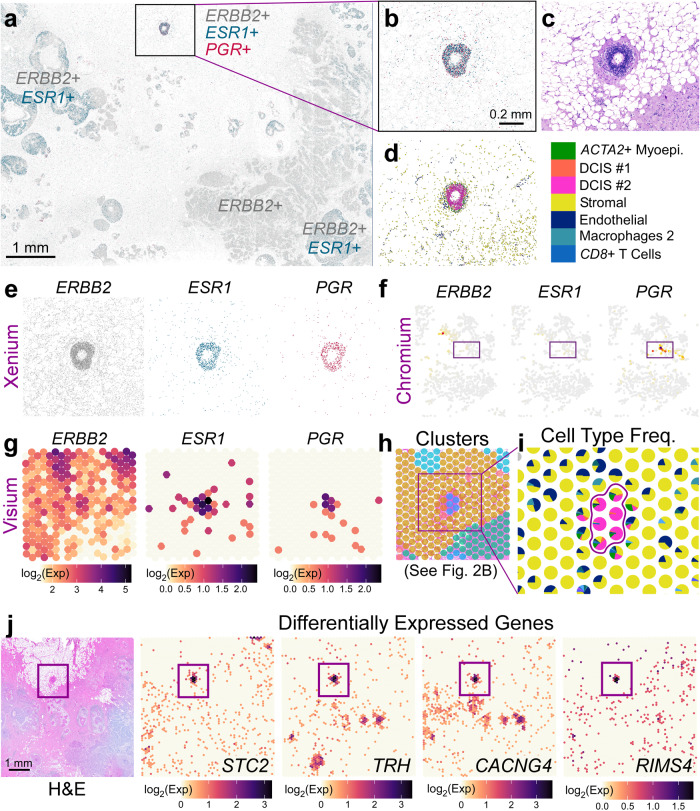
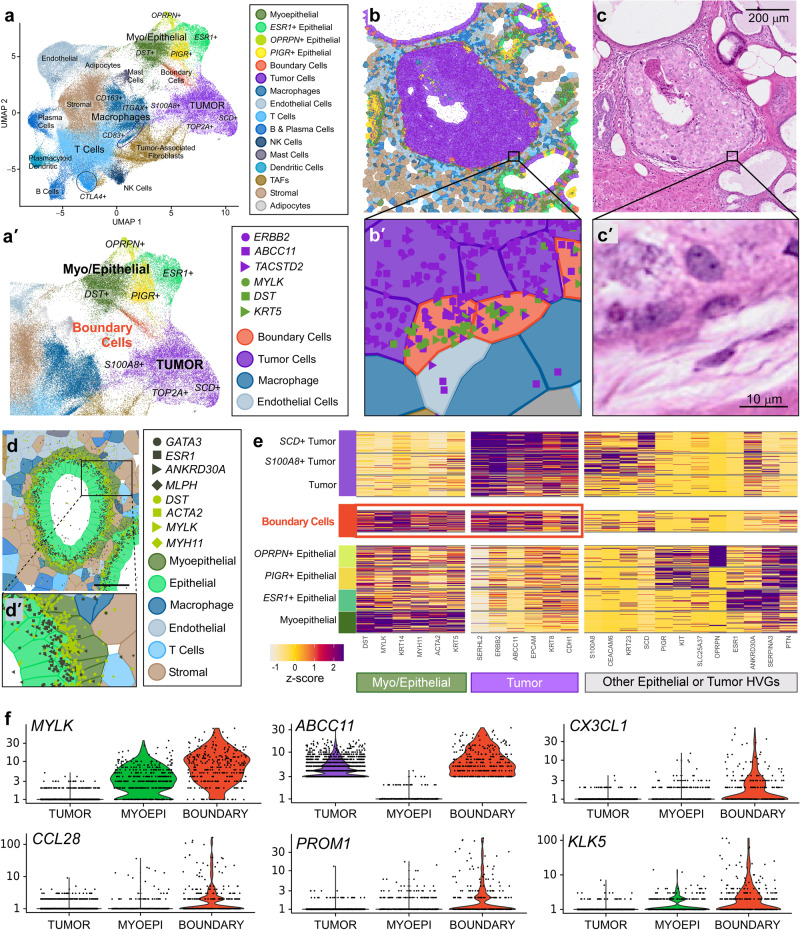
Single-cell and spatial technologies that profile gene expression across a whole tissue are revolutionizing the resolution of molecular states in clinical samples. Current commercially available technologies provide whole transcriptome single-cell, whole transcriptome spatial, or targeted in situ gene expression analysis. Here, we combine these technologies to explore tissue heterogeneity in large, FFPE human breast cancer sections. This integrative approach allowed us to explore molecular differences that exist between distinct tumor regions and to identify biomarkers involved in the progression towards invasive carcinoma. Further, we study cell neighborhoods and identify rare boundary cells that sit at the critical myoepithelial border confining the spread of malignant cells. Here, we demonstrate that each technology alone provides information about molecular signatures relevant to understanding cancer heterogeneity; however, it is the integration of these technologies that leads to deeper insights, ushering in discoveries that will progress oncology research and the development of diagnostics and therapeutics.
© 2023. The Author(s).
Conflict of interest statement
All authors are current or previous employees of 10x Genomics.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
