

Evaluation of noninvasive biospecimens for transcriptome studies
- PMID: 38114913
- PMCID: PMC10729488
- DOI: 10.1186/s12864-023-09875-4
Evaluation of noninvasive biospecimens for transcriptome studies
Abstract
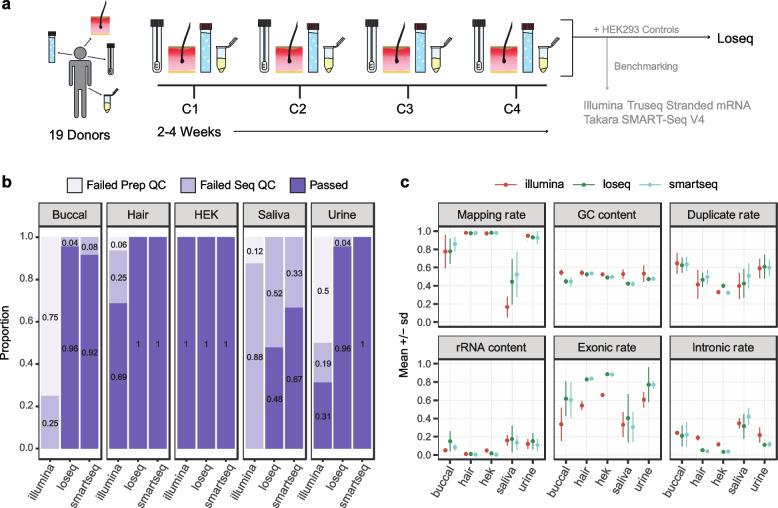
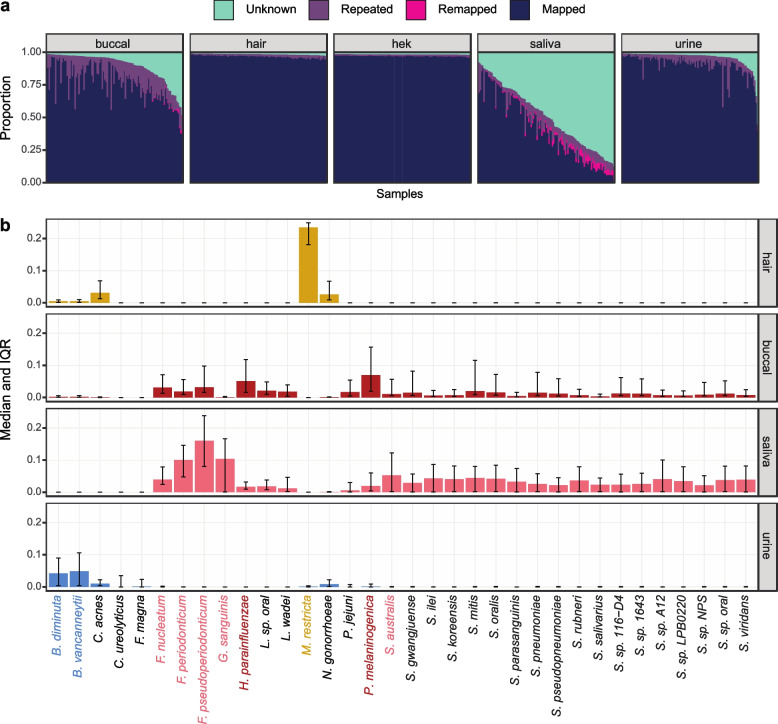
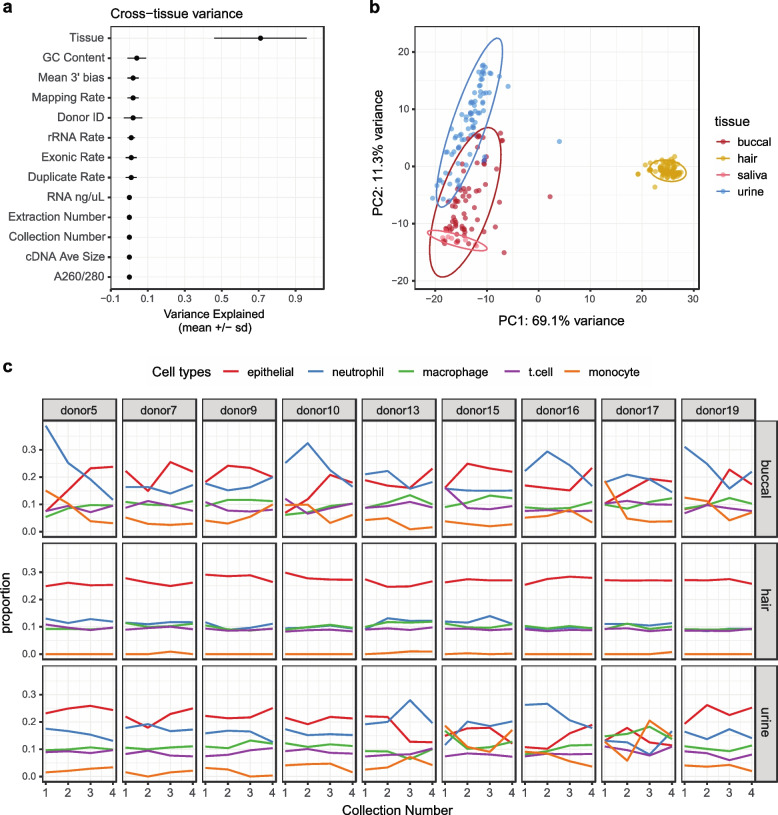
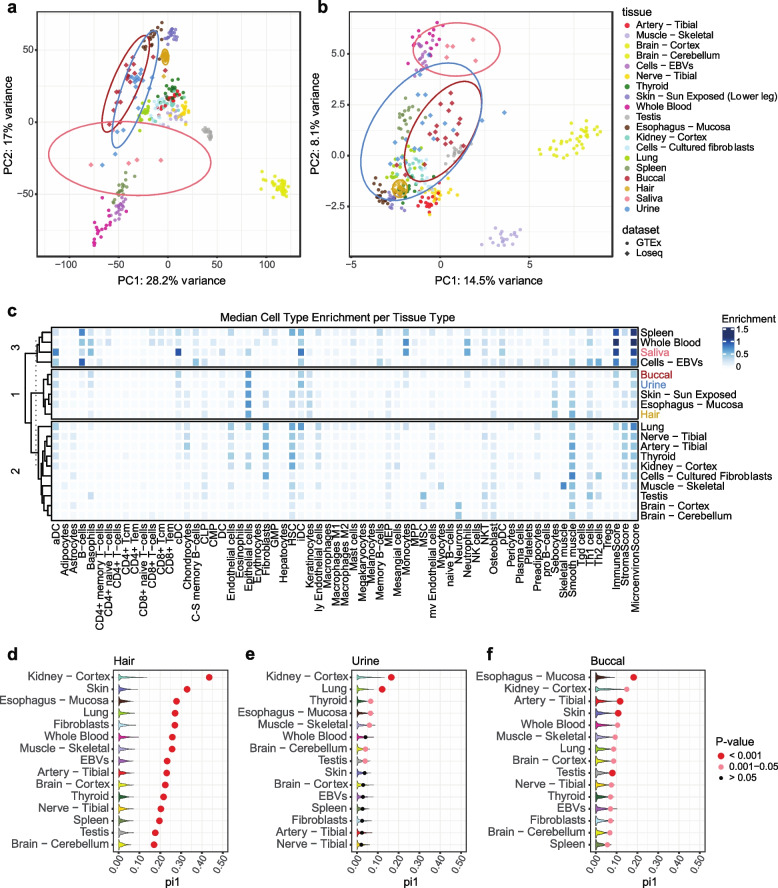
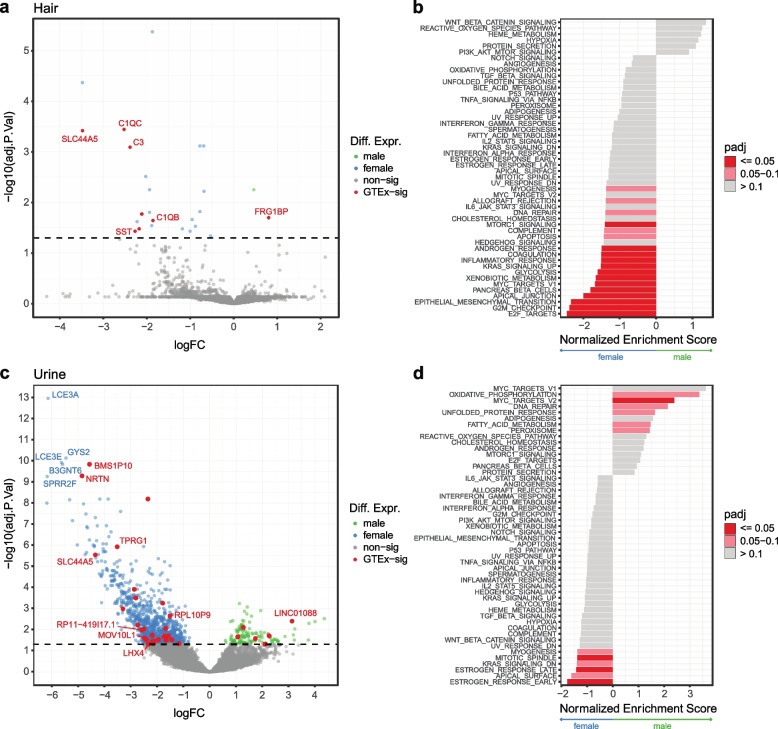
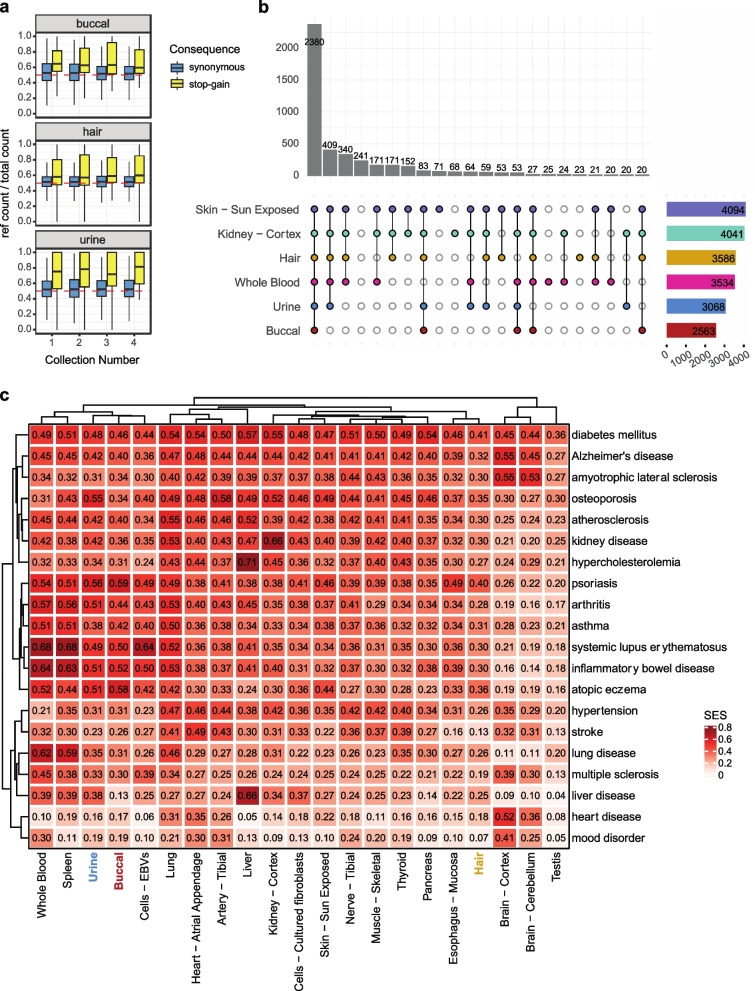
Transcriptome studies disentangle functional mechanisms of gene expression regulation and may elucidate the underlying biology of disease processes. However, the types of tissues currently collected typically assay a single post-mortem timepoint or are limited to investigating cell types found in blood. Noninvasive tissues may improve disease-relevant discovery by enabling more complex longitudinal study designs, by capturing different and potentially more applicable cell types, and by increasing sample sizes due to reduced collection costs and possible higher enrollment from vulnerable populations. Here, we develop methods for sampling noninvasive biospecimens, investigate their performance across commercial and in-house library preparations, characterize their biology, and assess the feasibility of using noninvasive tissues in a multitude of transcriptomic applications. We collected buccal swabs, hair follicles, saliva, and urine cell pellets from 19 individuals over three to four timepoints, for a total of 300 unique biological samples, which we then prepared with replicates across three library preparations, for a final tally of 472 transcriptomes. Of the four tissues we studied, we found hair follicles and urine cell pellets to be most promising due to the consistency of sample quality, the cell types and expression profiles we observed, and their performance in disease-relevant applications. This is the first study to thoroughly delineate biological and technical features of noninvasive samples and demonstrate their use in a wide array of transcriptomic and clinical analyses. We anticipate future use of these biospecimens will facilitate discovery and development of clinical applications.
Keywords: EQTLs; Hair follicles; Methods; Noninvasive RNA-sequencing; Transcriptomics; Urine cell pellets.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
