

Preleukemic single-cell landscapes reveal mutation-specific mechanisms and gene programs predictive of AML patient outcomes
- PMID: 38116120
- PMCID: PMC10726426
- DOI: 10.1016/j.xgen.2023.100426
Preleukemic single-cell landscapes reveal mutation-specific mechanisms and gene programs predictive of AML patient outcomes
Abstract
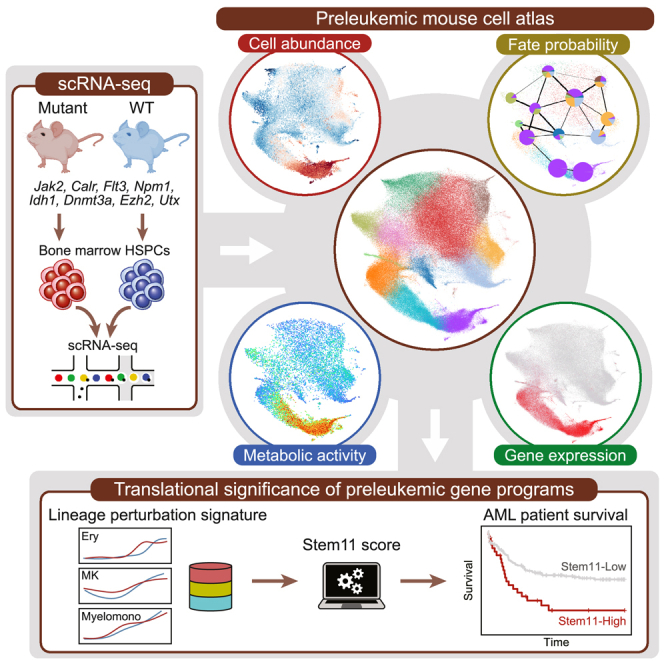
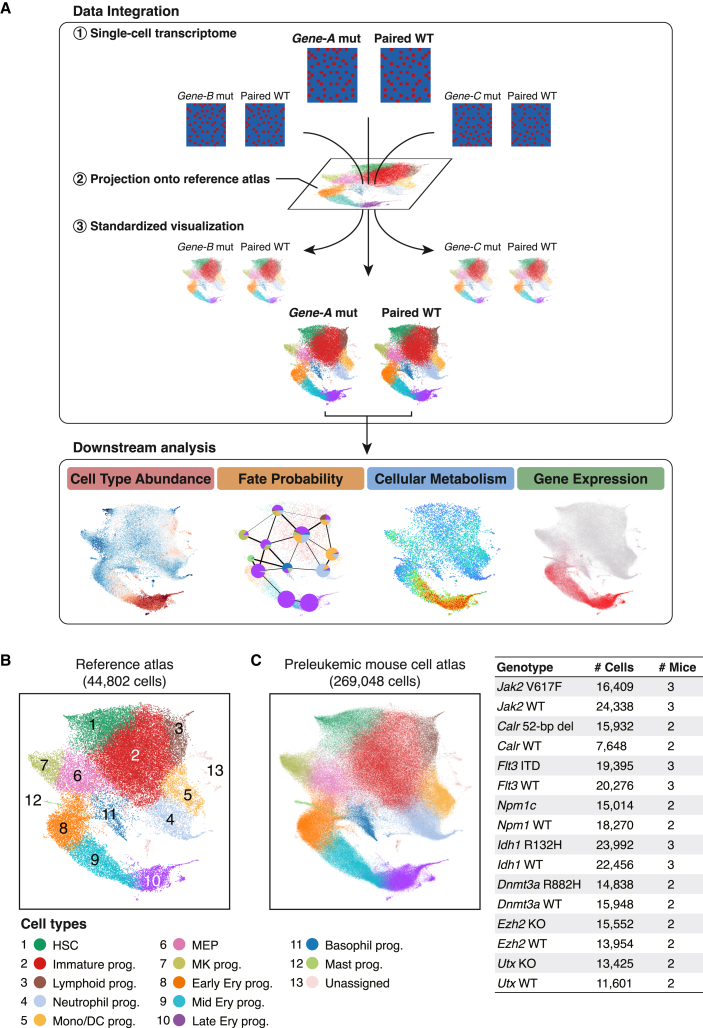
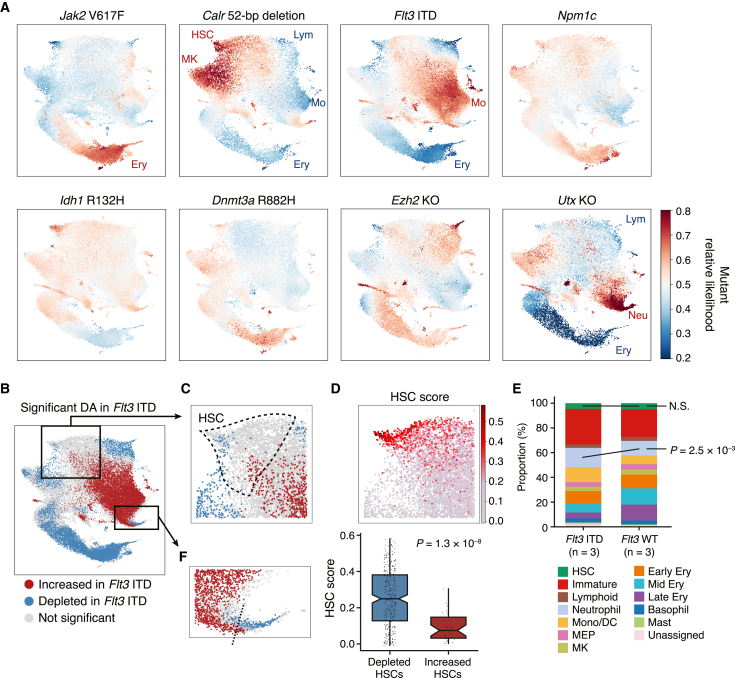
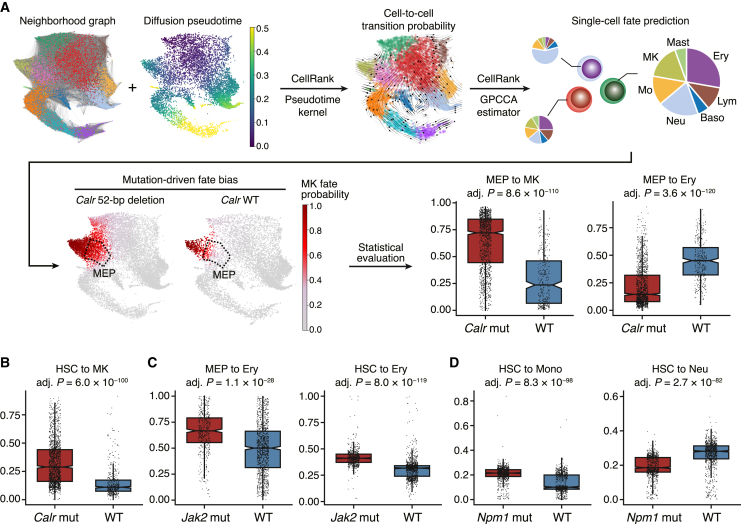
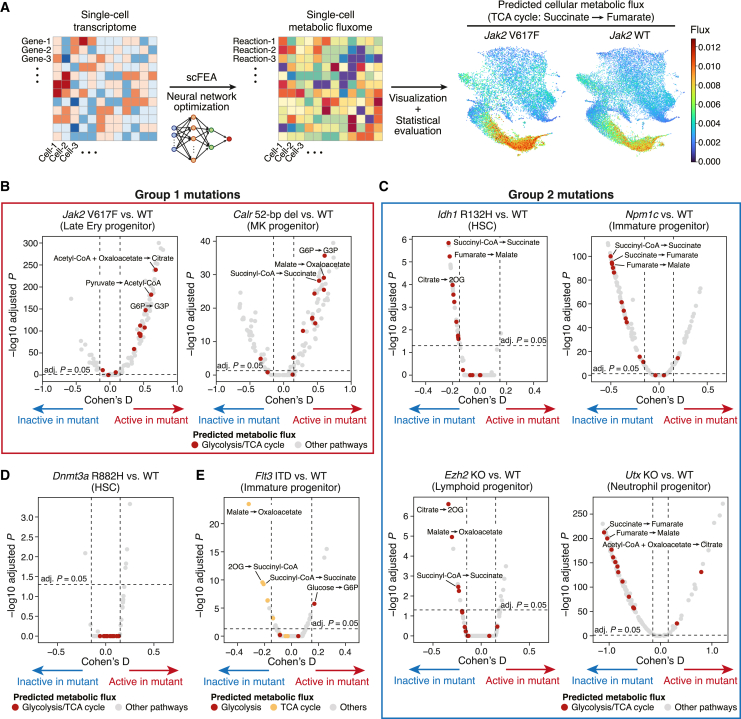
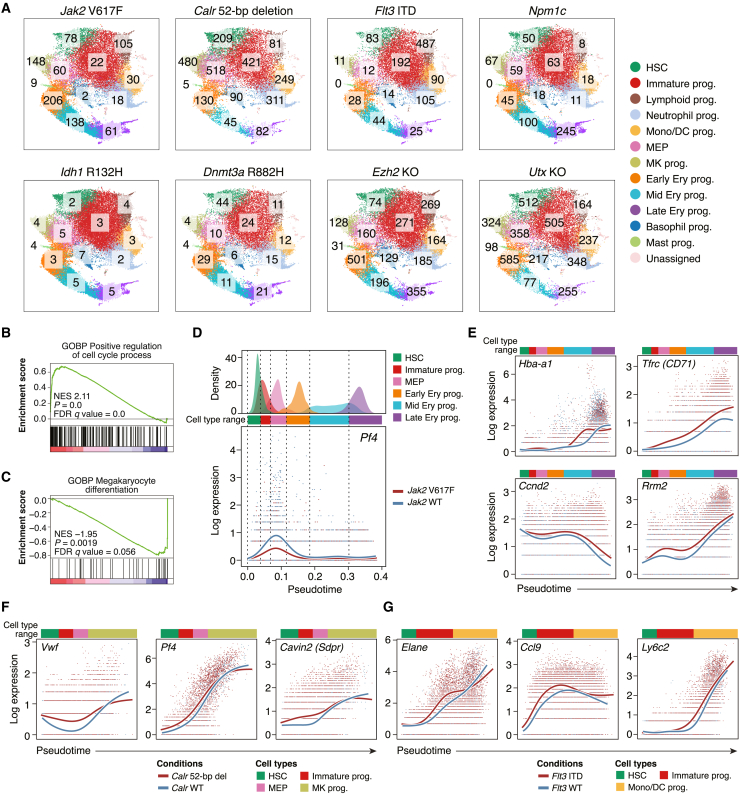
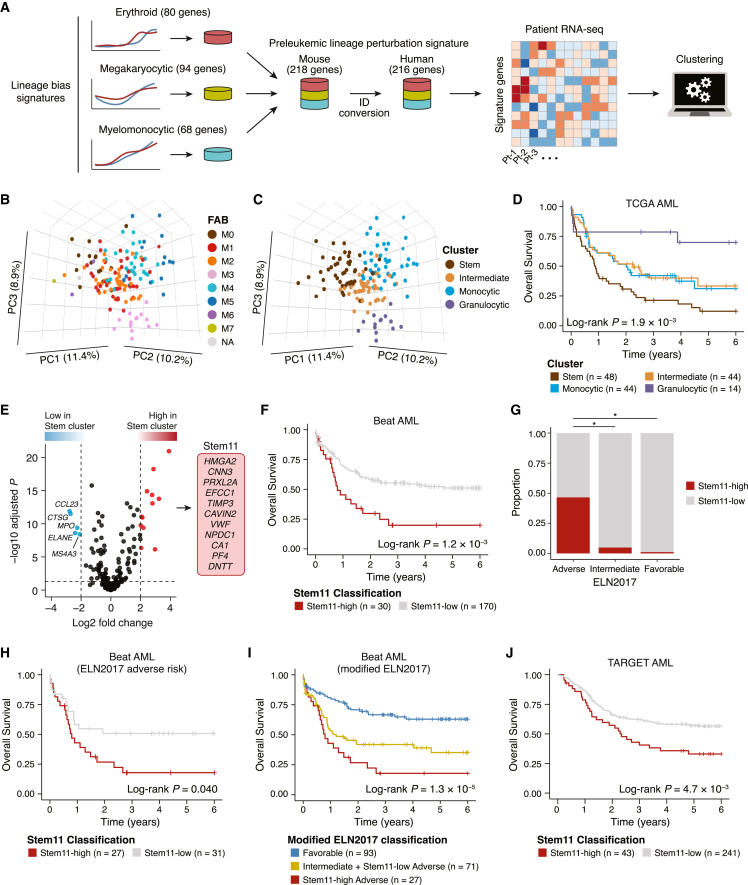
Acute myeloid leukemia (AML) and myeloid neoplasms develop through acquisition of somatic mutations that confer mutation-specific fitness advantages to hematopoietic stem and progenitor cells. However, our understanding of mutational effects remains limited to the resolution attainable within immunophenotypically and clinically accessible bulk cell populations. To decipher heterogeneous cellular fitness to preleukemic mutational perturbations, we performed single-cell RNA sequencing of eight different mouse models with driver mutations of myeloid malignancies, generating 269,048 single-cell profiles. Our analysis infers mutation-driven perturbations in cell abundance, cellular lineage fate, cellular metabolism, and gene expression at the continuous resolution, pinpointing cell populations with transcriptional alterations associated with differentiation bias. We further develop an 11-gene scoring system (Stem11) on the basis of preleukemic transcriptional signatures that predicts AML patient outcomes. Our results demonstrate that a single-cell-resolution deep characterization of preleukemic biology has the potential to enhance our understanding of AML heterogeneity and inform more effective risk stratification strategies.
Keywords: acute myeloid leukemia; hematopoiesis; myeloid malignancies; preleukemia; single-cell RNA-seq.
© 2023 The Author(s).
Conflict of interest statement
Aspects of this work are included in United Kingdom patent application 2312684.0.
Figures
References
-
- Granfeldt Østgård L.S., Medeiros B.C., Sengeløv H., Nørgaard M., Andersen M.K., Dufva I.H., Friis L.S., Kjeldsen E., Marcher C.W., Preiss B., et al. Epidemiology and Clinical Significance of Secondary and Therapy-Related Acute Myeloid Leukemia: A National Population-Based Cohort Study. J. Clin. Oncol. 2015;33:3641–3649. doi: 10.1200/jco.2014.60.0890. - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
