

Clinical and Genotype Characteristics and Symptom Migration in Patients With Mixed Phenotype Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey
- PMID: 38117424
- PMCID: PMC10899146
- DOI: 10.1007/s40119-023-00344-3
Clinical and Genotype Characteristics and Symptom Migration in Patients With Mixed Phenotype Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey
Abstract
Introduction: Transthyretin amyloidosis (ATTR amyloidosis) is primarily associated with a cardiac or neurologic phenotype, but a mixed phenotype is increasingly described.
Methods: This study describes the mixed phenotype cohort in the Transthyretin Amyloidosis Outcomes Survey (THAOS). THAOS is an ongoing, longitudinal, observational survey of patients with ATTR amyloidosis, including both hereditary (ATTRv) and wild-type disease, and asymptomatic carriers of pathogenic transthyretin variants. Baseline characteristics of patients with a mixed phenotype (at enrollment or reclassified during follow-up) are described (data cutoff: January 4, 2022).
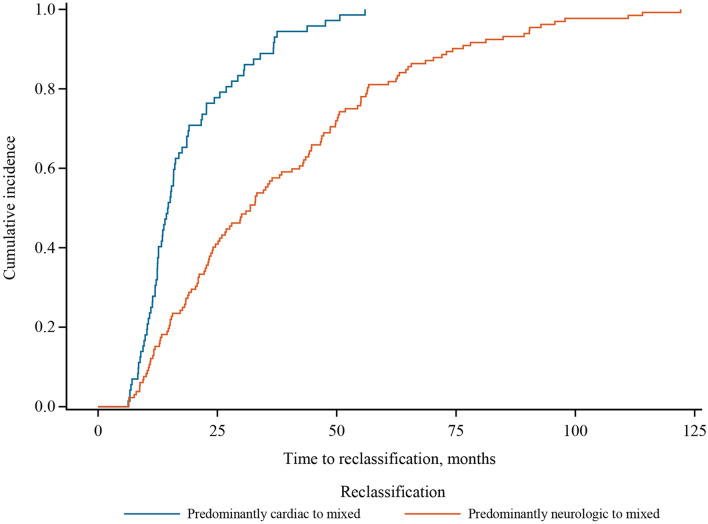
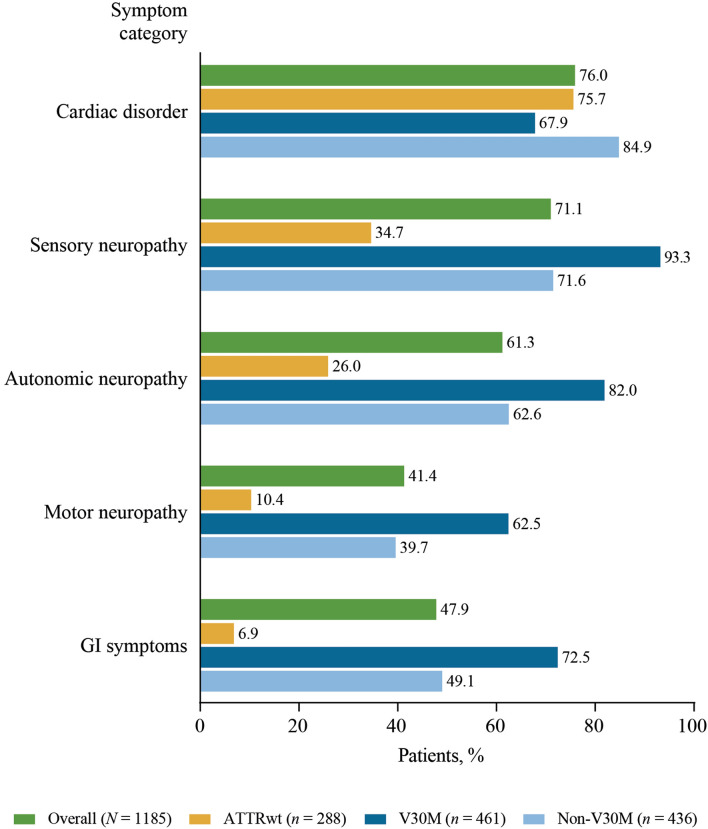
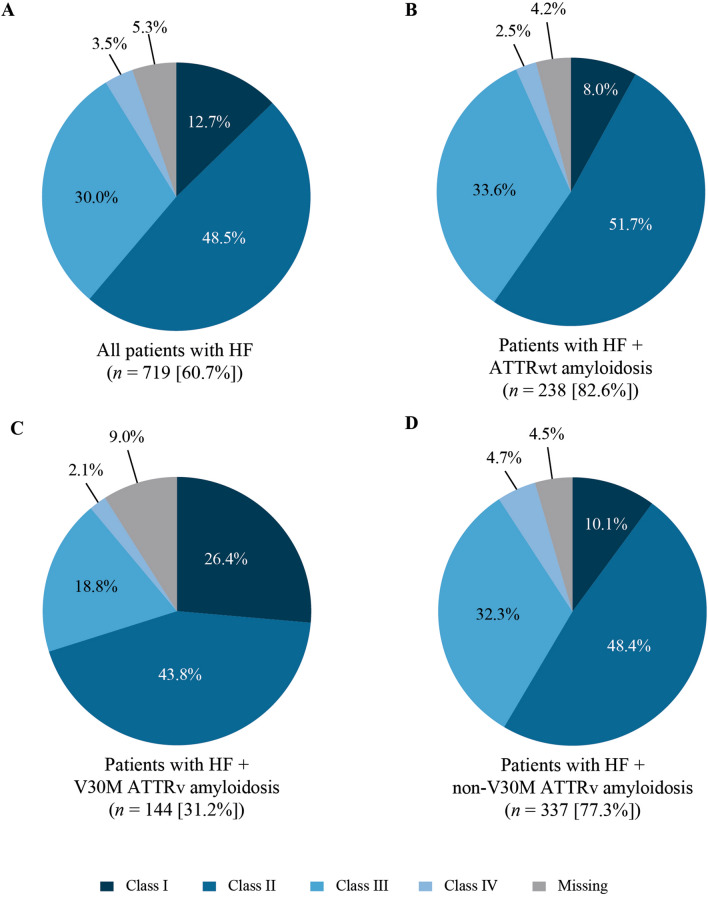
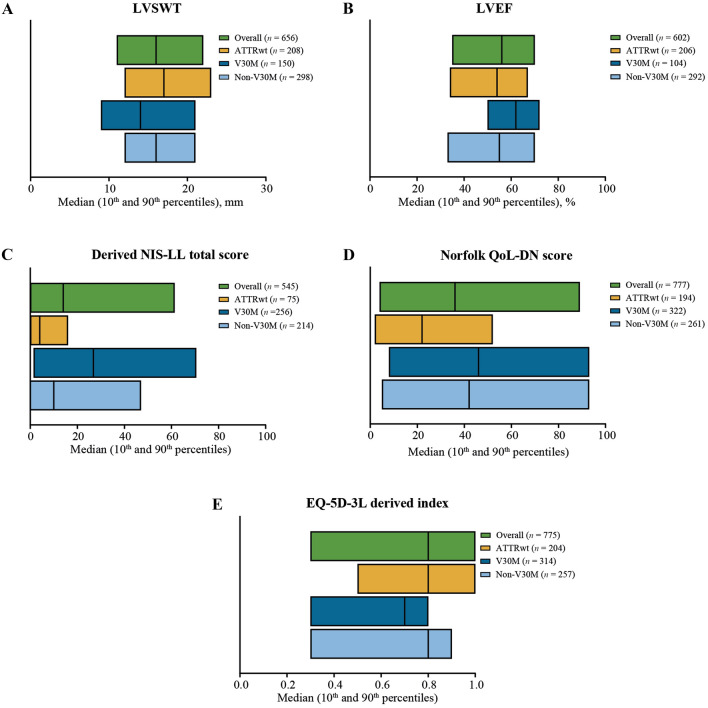
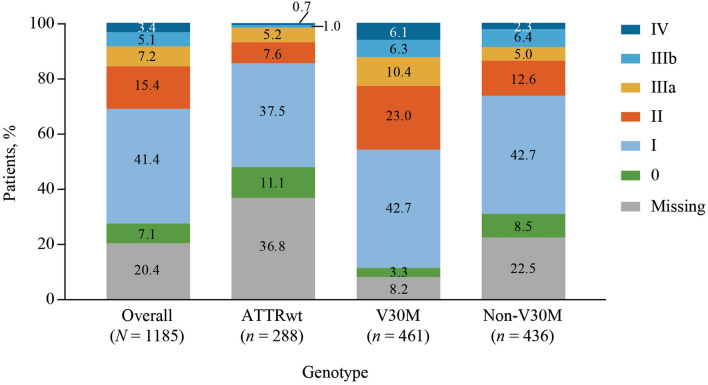
Results: Approximately one-third of symptomatic patients (n = 1185/3542; 33.5%) were classified at enrollment or follow-up as mixed phenotype (median age, 66.5 years). Of those, 344 (29.0%) were reclassified to mixed phenotype within a median 1-2 years of follow-up. Most patients with mixed phenotype had ATTRv amyloidosis (75.7%). The most frequent genotypes were V30M (38.9%) and wild type (24.3%).
Conclusions: These THAOS data represent the largest analysis of a real-world mixed phenotype ATTR amyloidosis population to date and suggest that a mixed phenotype may be more prevalent than previously thought. Patients may also migrate from a primarily neurologic or cardiologic presentation to a mixed phenotype over time. These data reinforce the need for multidisciplinary evaluation at initial assessment and follow-up of all patients with ATTR amyloidosis.
Trial registration: ClinicalTrials.gov: NCT00628745.
Keywords: Amyloidosis; Cardiomyopathy; Mixed phenotype; Polyneuropathy; THAOS; Transthyretin.
© 2023. The Author(s).
Conflict of interest statement
Juan González-Moreno reports speaker fees from Akcea, Alnylam, and Pfizer. Angela Dispenzieri reports research grants from Alnylam, Celgene, Janssen, Millennium, and Pfizer; funding for meeting expenses (travel) from Pfizer; and advisory board fees from Akcea and Intellia. Martha Grogan reports grants and advisory board/consultancy fees paid to her institution from Alnylam, Eidos, Pfizer, and Prothena. Teresa Coelho served as a medical advisor for Pfizer, without honoraria; and reports funding for meeting expenses (travel, accommodation, registration) from Pfizer. Ivailo Tournev reports research funding, honoraria for presentations, and advisory board fees from Alnylam, Pfizer, and Pharma Pro. Márcia Waddington-Cruz reports research funding, consulting fees, and travel support for advisory boards and meetings from FoldRx Pharmaceuticals and Pfizer. Jonas Wixner reports consulting fees and travel support for lectures and advisory boards from Alnylam and Pfizer; and consulting fees from Akcea. Igor Diemberger reports research funding and speaker fees from Daiichi Sankyo and Pfizer (outside scope of present research). Pablo Garcia-Pavia reports speaker fees from Akcea, Alnylam, Eidos, and Pfizer; consulting fees from Alnylam, Akcea, AstraZeneca, Eidos, Neurimmune, and Pfizer; and research/educational support to his institution from Alnylam, Eidos, and Pfizer. Doug Chapman, Pritam Gupta, Oliver Glass, and Leslie Amass are full-time employees of Pfizer and hold stock and/or stock options.
Figures
References
-
- Rowczenio D, Wechalekar A. Mutations in hereditary amyloidosis 2015. Available from: http://amyloidosismutations.com/mut-attr.php Accessed 1 June 2023.
-
- Dispenzieri A, Coelho T, Conceicao I, Waddington-Cruz M, Wixner J, Kristen AV, et al. Clinical and genetic profile of patients enrolled in the Transthyretin Amyloidosis Outcomes Survey (THAOS): 14-year update. Orphanet J Rare Dis. 2022;17(1):236. doi: 10.1186/s13023-022-02359-w. - DOI - PMC - PubMed
Associated data
LinkOut - more resources
Full Text Sources
Medical
Research Materials
