

Bi-allelic genetic variants in the translational GTPases GTPBP1 and GTPBP2 cause a distinct identical neurodevelopmental syndrome
- PMID: 38118446
- PMCID: PMC10806450
- DOI: 10.1016/j.ajhg.2023.11.012
Bi-allelic genetic variants in the translational GTPases GTPBP1 and GTPBP2 cause a distinct identical neurodevelopmental syndrome
Abstract
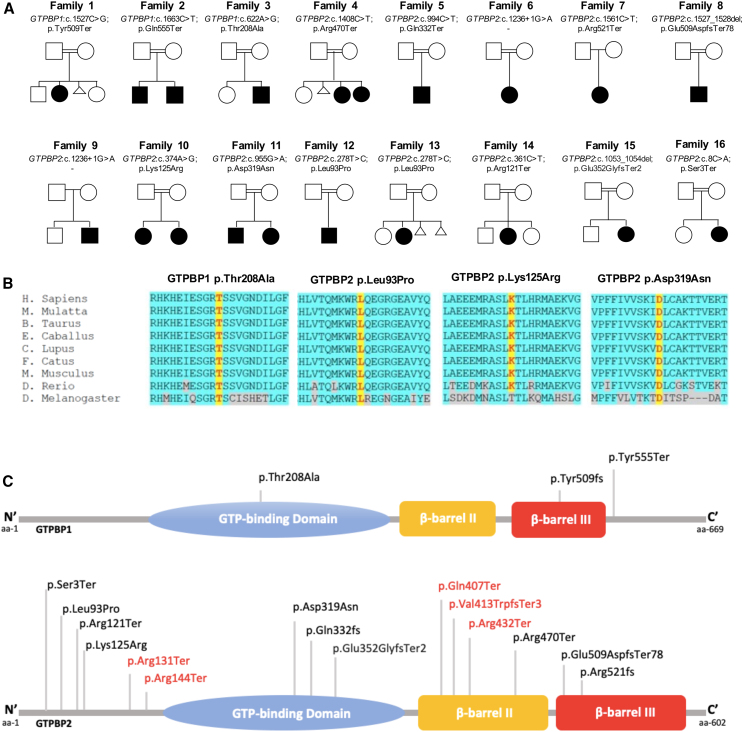
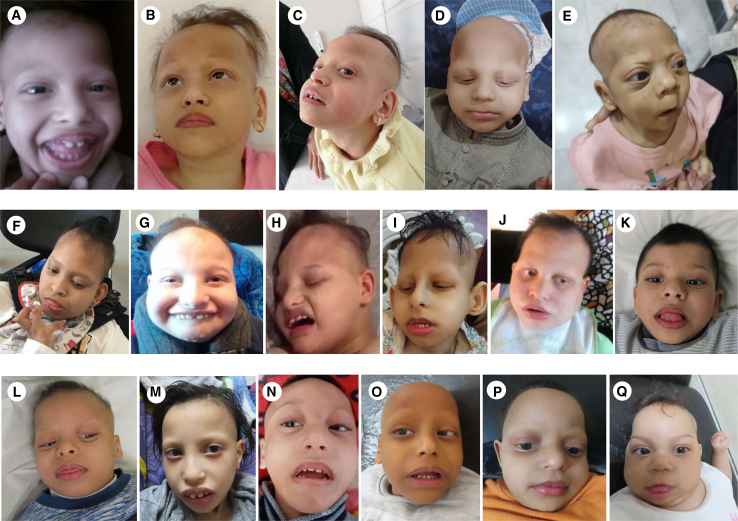
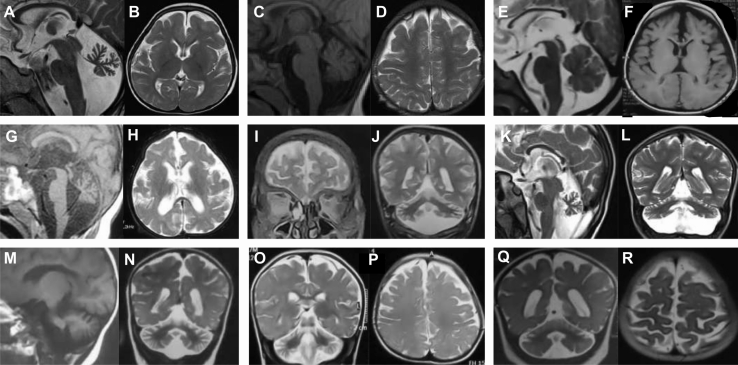
The homologous genes GTPBP1 and GTPBP2 encode GTP-binding proteins 1 and 2, which are involved in ribosomal homeostasis. Pathogenic variants in GTPBP2 were recently shown to be an ultra-rare cause of neurodegenerative or neurodevelopmental disorders (NDDs). Until now, no human phenotype has been linked to GTPBP1. Here, we describe individuals carrying bi-allelic GTPBP1 variants that display an identical phenotype with GTPBP2 and characterize the overall spectrum of GTP-binding protein (1/2)-related disorders. In this study, 20 individuals from 16 families with distinct NDDs and syndromic facial features were investigated by whole-exome (WES) or whole-genome (WGS) sequencing. To assess the functional impact of the identified genetic variants, semi-quantitative PCR, western blot, and ribosome profiling assays were performed in fibroblasts from affected individuals. We also investigated the effect of reducing expression of CG2017, an ortholog of human GTPBP1/2, in the fruit fly Drosophila melanogaster. Individuals with bi-allelic GTPBP1 or GTPBP2 variants presented with microcephaly, profound neurodevelopmental impairment, pathognomonic craniofacial features, and ectodermal defects. Abnormal vision and/or hearing, progressive spasticity, choreoathetoid movements, refractory epilepsy, and brain atrophy were part of the core phenotype of this syndrome. Cell line studies identified a loss-of-function (LoF) impact of the disease-associated variants but no significant abnormalities on ribosome profiling. Reduced expression of CG2017 isoforms was associated with locomotor impairment in Drosophila. In conclusion, bi-allelic GTPBP1 and GTPBP2 LoF variants cause an identical, distinct neurodevelopmental syndrome. Mutant CG2017 knockout flies display motor impairment, highlighting the conserved role for GTP-binding proteins in CNS development across species.
Keywords: GREND syndrome; GTPBP1; GTPBP2; NBIA; animal models; ectodermal disorders; neurodegeneration; neurodevelopmental disorders; ribosome stalling; ribosomopathies.
Copyright © 2023 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures


References
-
- Joazeiro C.A.P. Ribosomal stalling during translation: providing substrates for ribosome-associated protein quality control. Annu. Rev. Cell Dev. Biol. 2017;33:343–368. - PubMed
-
- Jaberi E., Rohani M., Shahidi G.A., Nafissi S., Arefian E., Soleimani M., Rasooli P., Ahmadieh H., Daftarian N., Carrami E.M., et al. Identification of mutation in GTPBP2 in patients of a family with neurodegeneration accompanied by iron deposition in the brain. Neurobiol. Aging. 2016;38:216-e11. - PubMed
-
- Bertoli-Avella A.M., Garcia-Aznar J.M., Brandau O., Al-Hakami F., Yüksel Z., Marais A., Grüning N.M., Abbasi Moheb L., Paknia O., Alshaikh N., et al. Biallelic inactivating variants in the GTPBP2 gene cause a neurodevelopmental disorder with severe intellectual disability. Eur. J. Hum. Genet. 2018;26:592–598. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
