

Multiomic spatial landscape of innate immune cells at human central nervous system borders
- PMID: 38123840
- PMCID: PMC10803260
- DOI: 10.1038/s41591-023-02673-1
Multiomic spatial landscape of innate immune cells at human central nervous system borders
Abstract
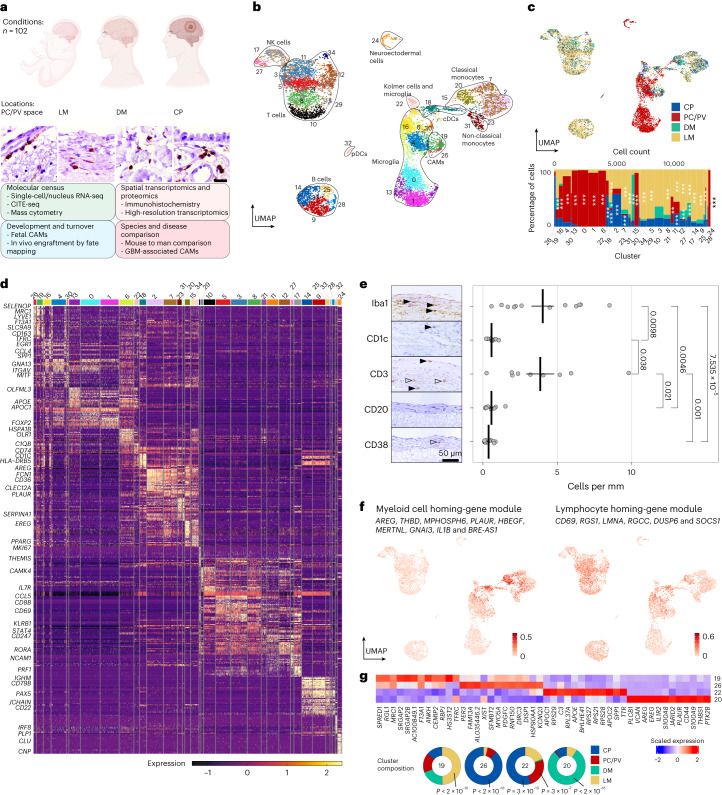
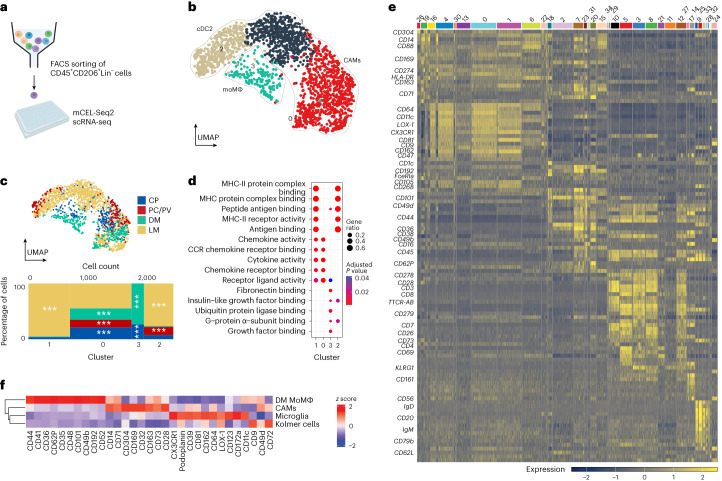
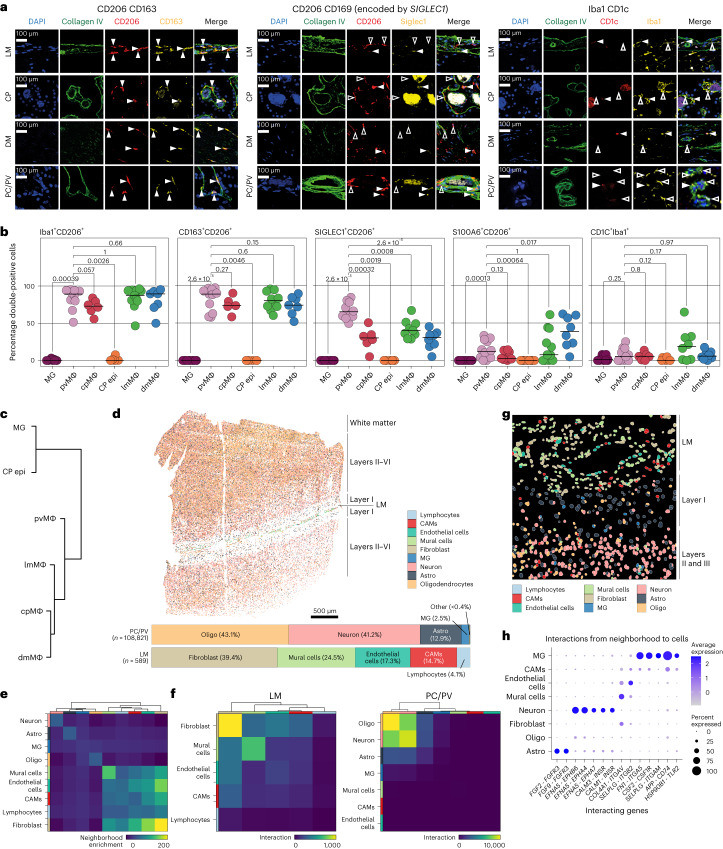
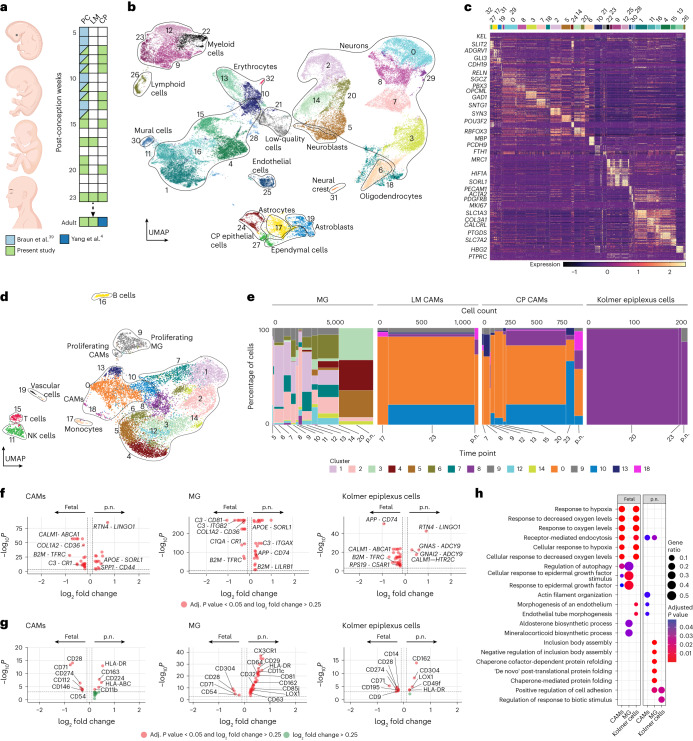
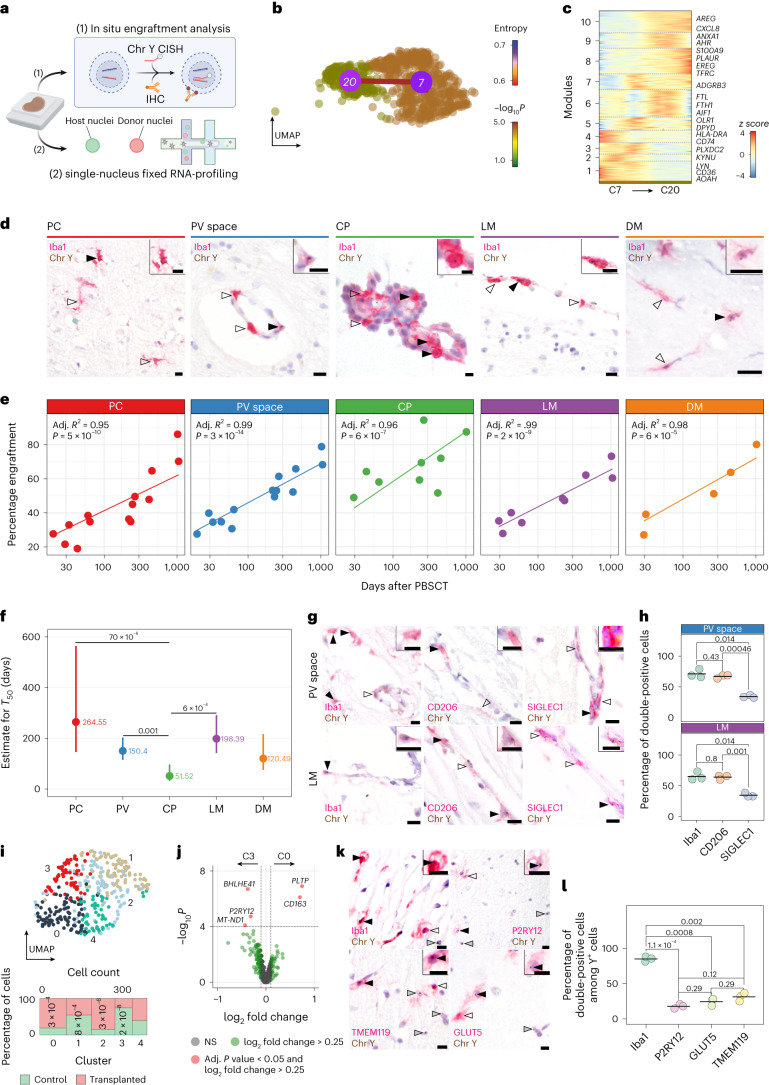
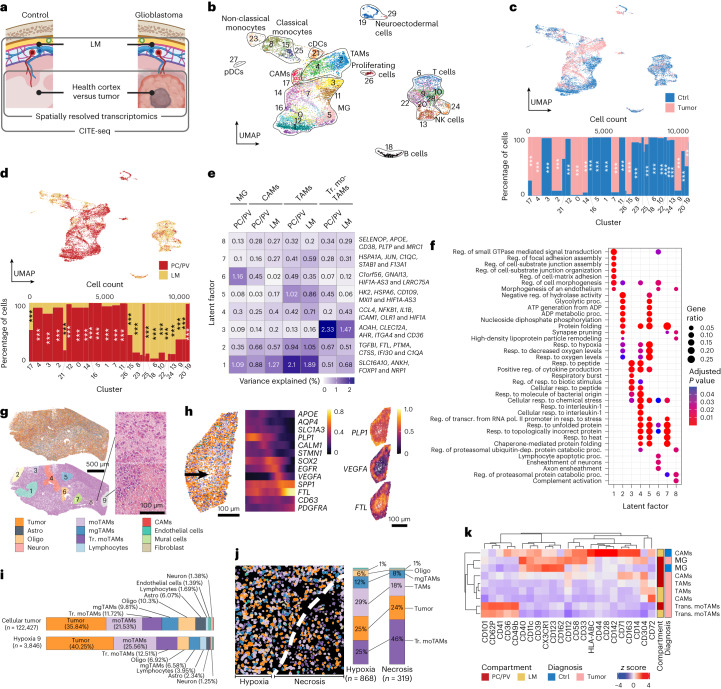
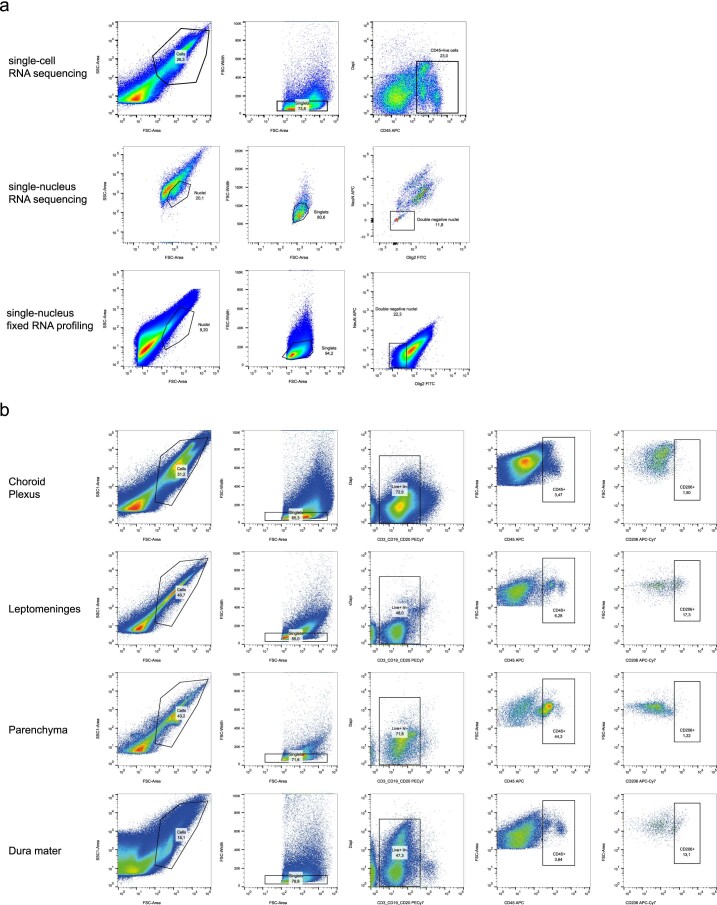
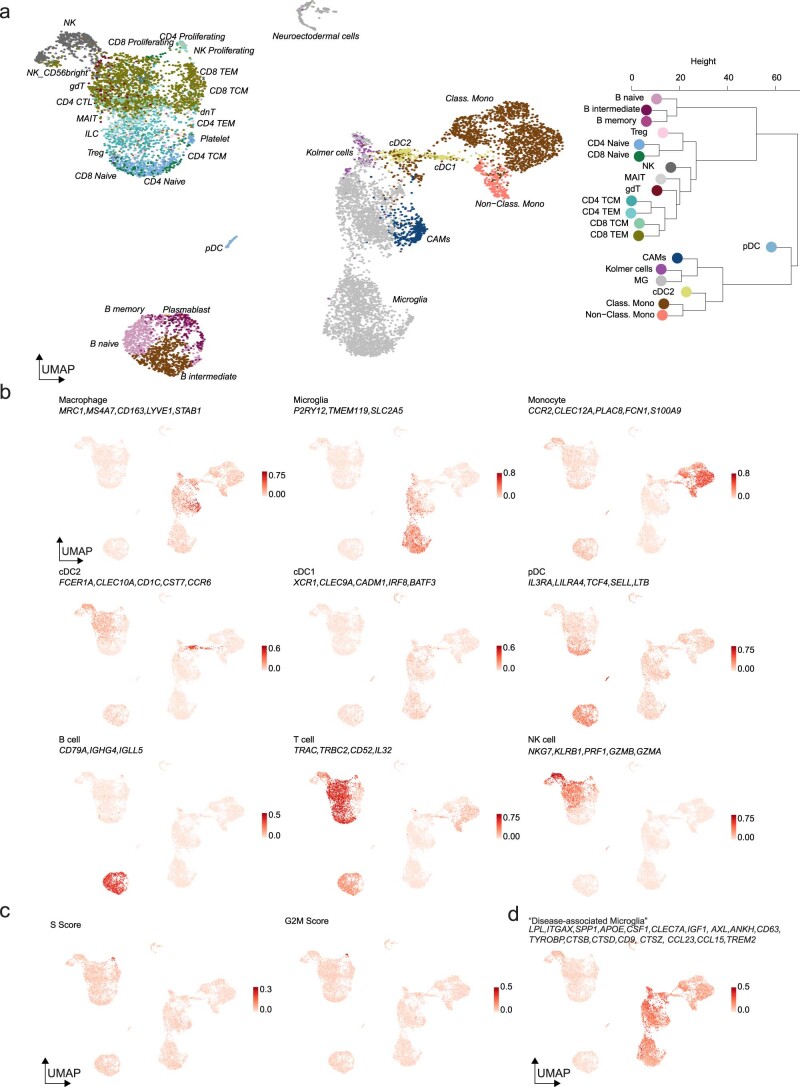
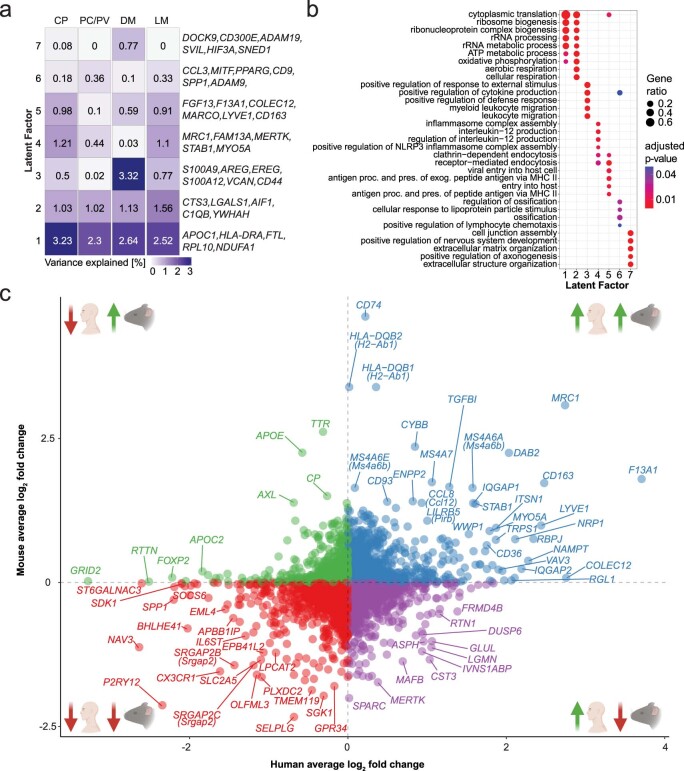
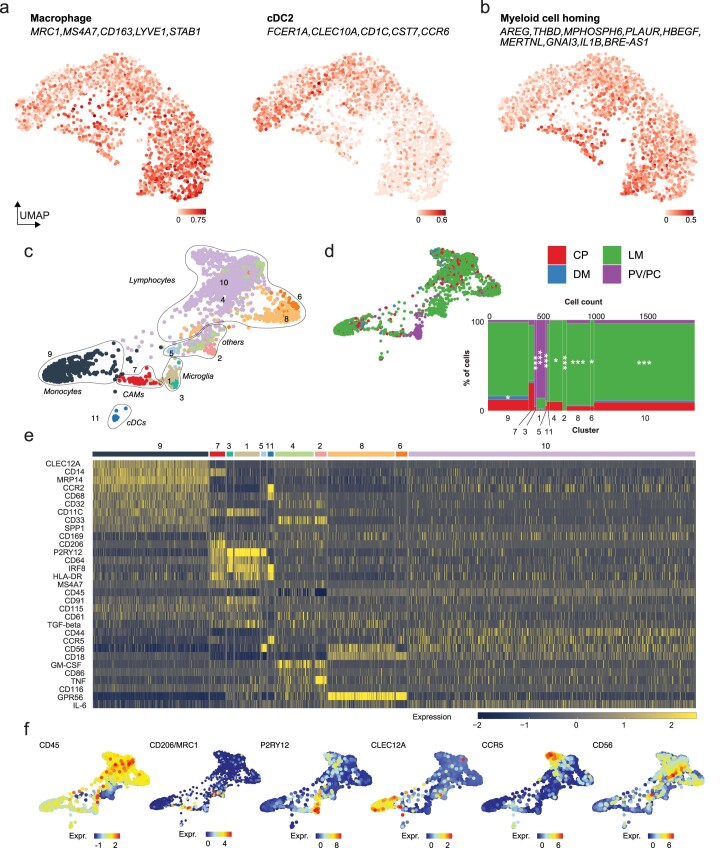
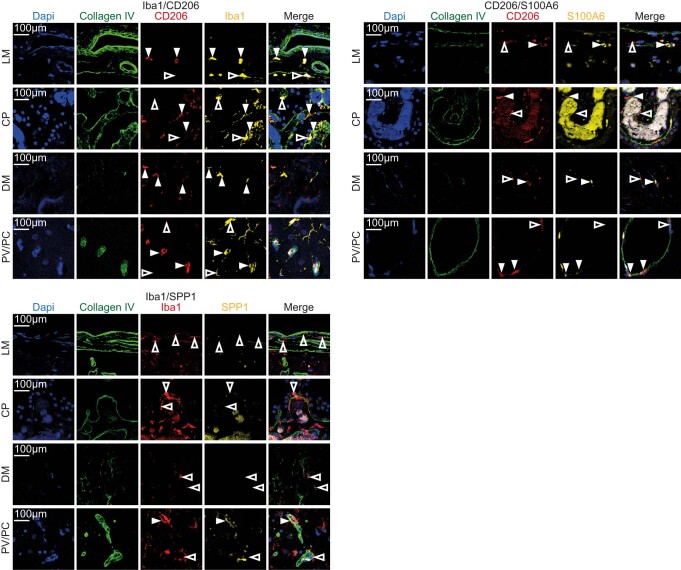
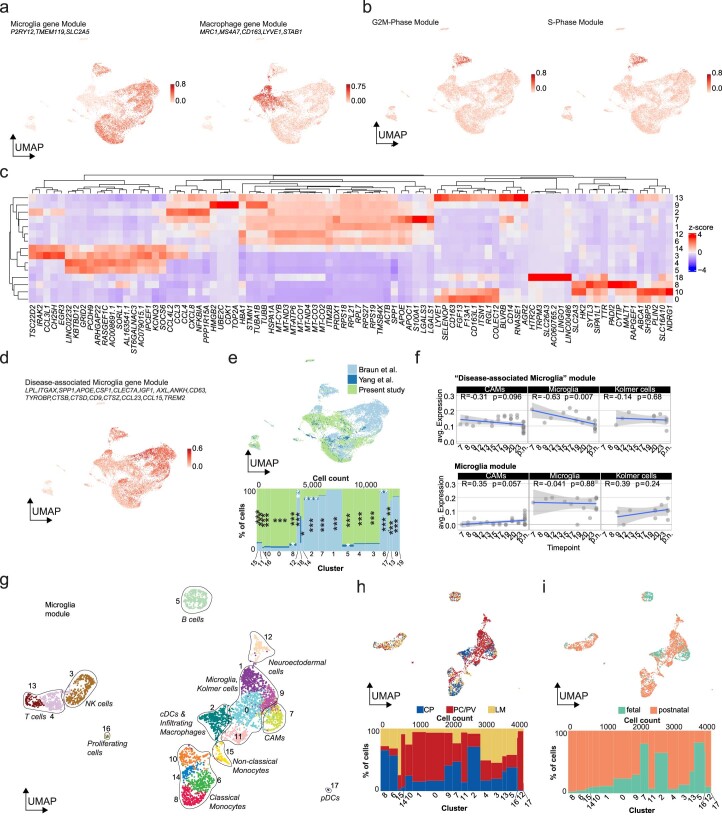
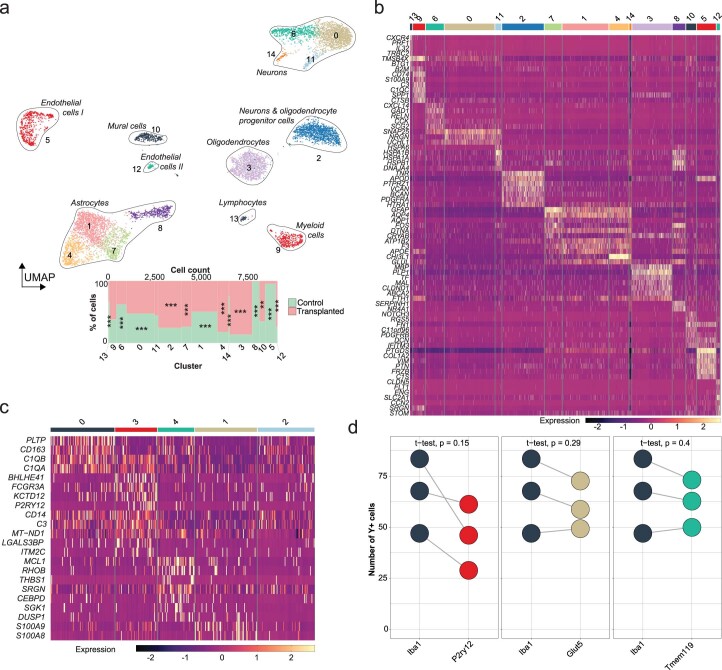
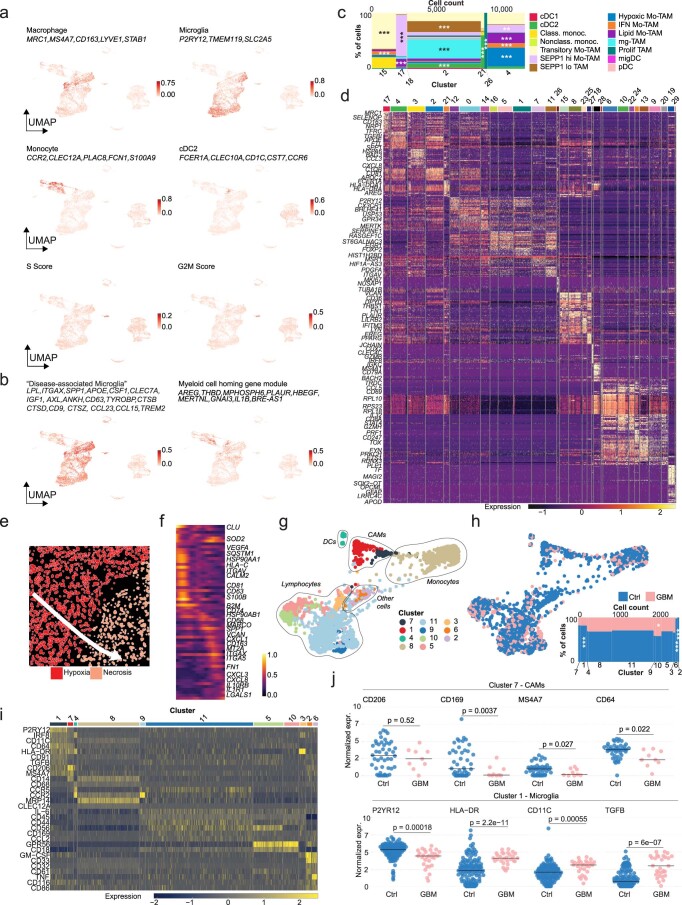
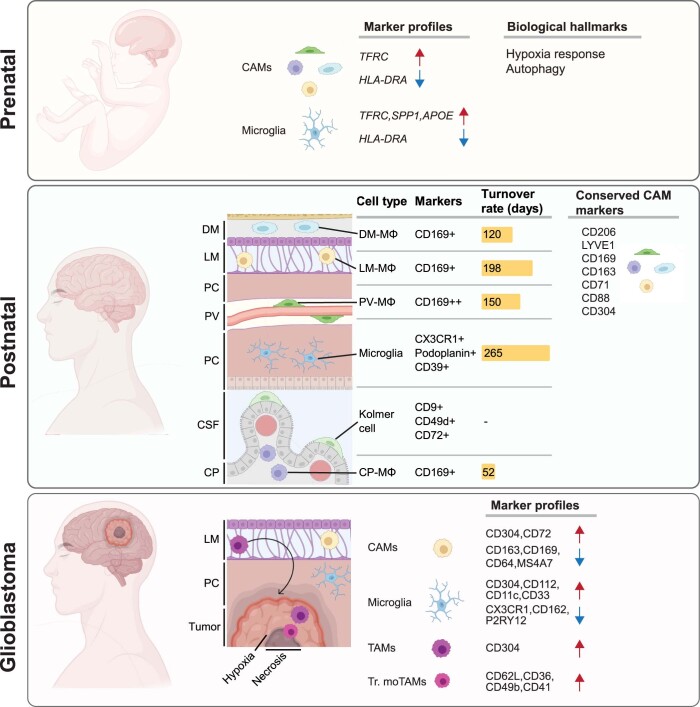
The innate immune compartment of the human central nervous system (CNS) is highly diverse and includes several immune-cell populations such as macrophages that are frequent in the brain parenchyma (microglia) and less numerous at the brain interfaces as CNS-associated macrophages (CAMs). Due to their scantiness and particular location, little is known about the presence of temporally and spatially restricted CAM subclasses during development, health and perturbation. Here we combined single-cell RNA sequencing, time-of-flight mass cytometry and single-cell spatial transcriptomics with fate mapping and advanced immunohistochemistry to comprehensively characterize the immune system at human CNS interfaces with over 356,000 analyzed transcriptomes from 102 individuals. We also provide a comprehensive analysis of resident and engrafted myeloid cells in the brains of 15 individuals with peripheral blood stem cell transplantation, revealing compartment-specific engraftment rates across different CNS interfaces. Integrated multiomic and high-resolution spatial transcriptome analysis of anatomically dissected glioblastoma samples shows regionally distinct myeloid cell-type distributions driven by hypoxia. Notably, the glioblastoma-associated hypoxia response was distinct from the physiological hypoxia response in fetal microglia and CAMs. Our results highlight myeloid diversity at the interfaces of the human CNS with the periphery and provide insights into the complexities of the human brain's immune system.
© 2023. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
