

GLP-1 metabolite GLP-1(9-36) is a systemic inhibitor of mouse and human pancreatic islet glucagon secretion
- PMID: 38127123
- PMCID: PMC10844371
- DOI: 10.1007/s00125-023-06060-w
GLP-1 metabolite GLP-1(9-36) is a systemic inhibitor of mouse and human pancreatic islet glucagon secretion
Abstract
Aims/hypothesis: Diabetes mellitus is associated with impaired insulin secretion, often aggravated by oversecretion of glucagon. Therapeutic interventions should ideally correct both defects. Glucagon-like peptide 1 (GLP-1) has this capability but exactly how it exerts its glucagonostatic effect remains obscure. Following its release GLP-1 is rapidly degraded from GLP-1(7-36) to GLP-1(9-36). We hypothesised that the metabolite GLP-1(9-36) (previously believed to be biologically inactive) exerts a direct inhibitory effect on glucagon secretion and that this mechanism becomes impaired in diabetes.
Methods: We used a combination of glucagon secretion measurements in mouse and human islets (including islets from donors with type 2 diabetes), total internal reflection fluorescence microscopy imaging of secretory granule dynamics, recordings of cytoplasmic Ca2+ and measurements of protein kinase A activity, immunocytochemistry, in vivo physiology and GTP-binding protein dissociation studies to explore how GLP-1 exerts its inhibitory effect on glucagon secretion and the role of the metabolite GLP-1(9-36).
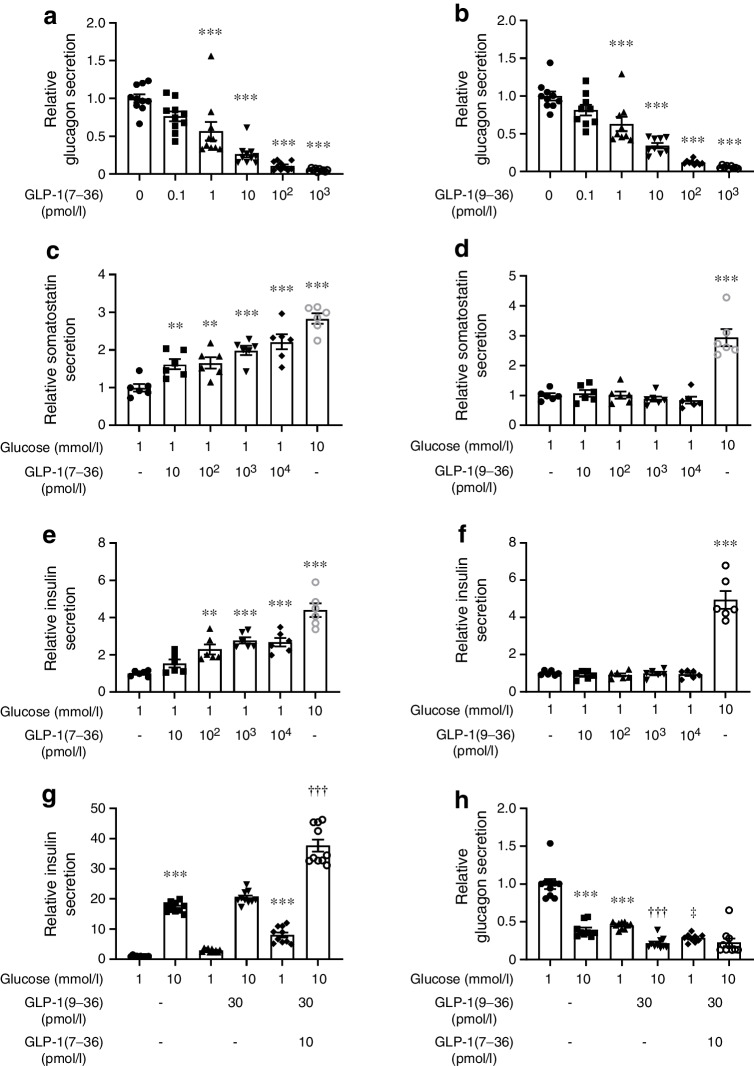
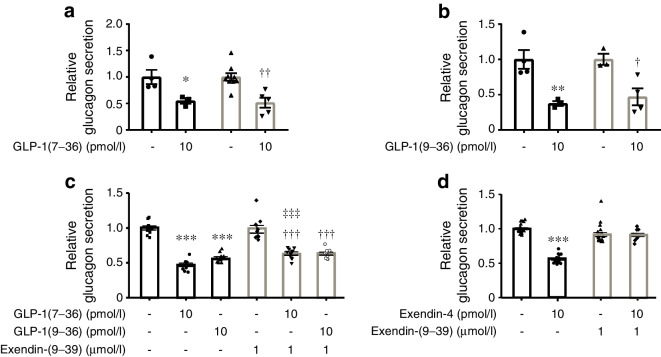
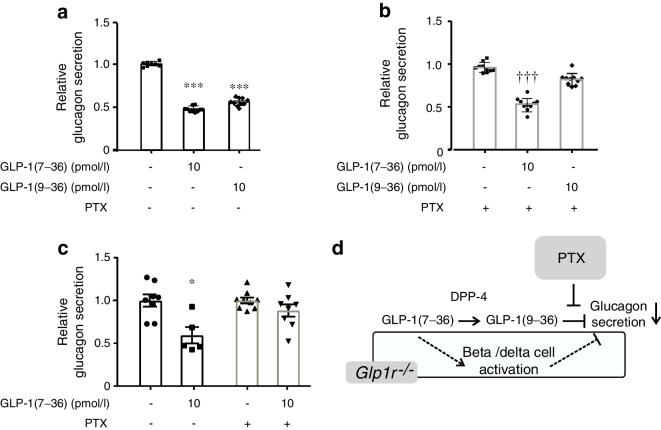
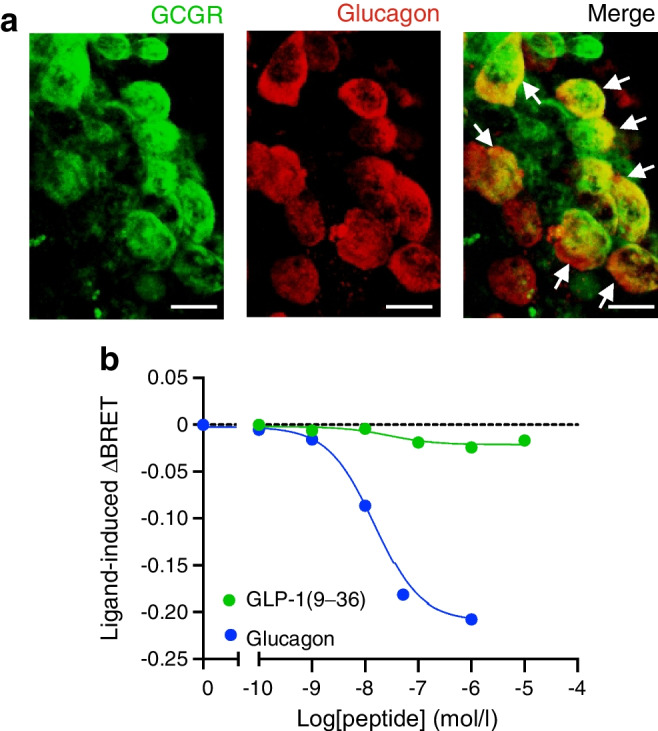
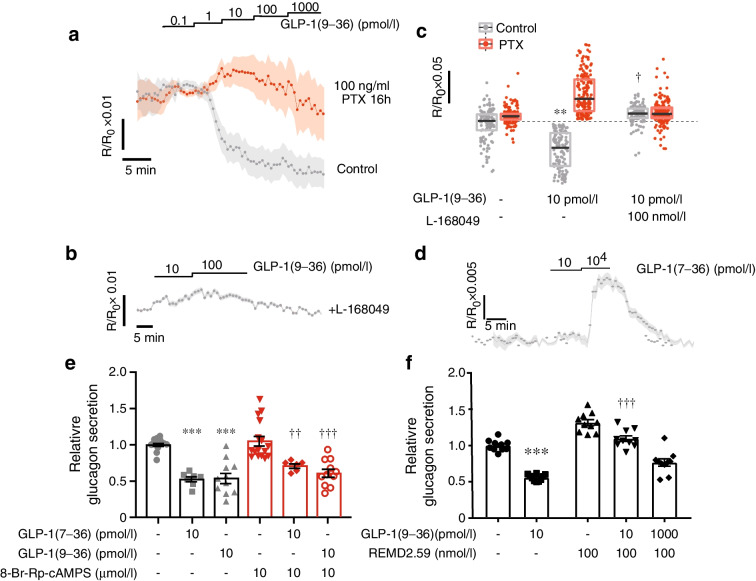
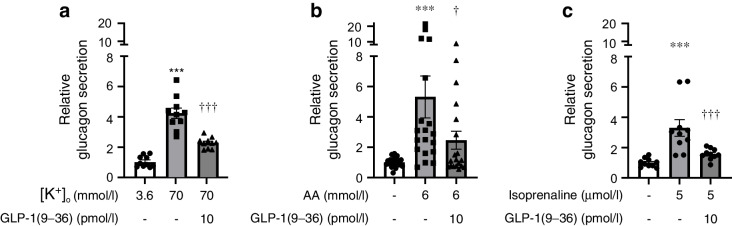
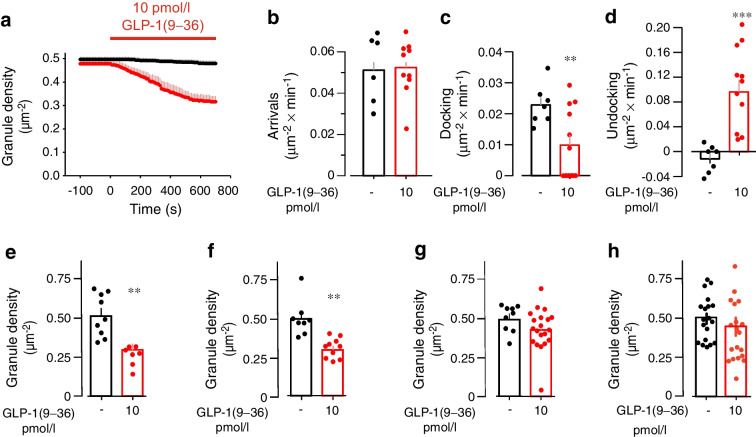
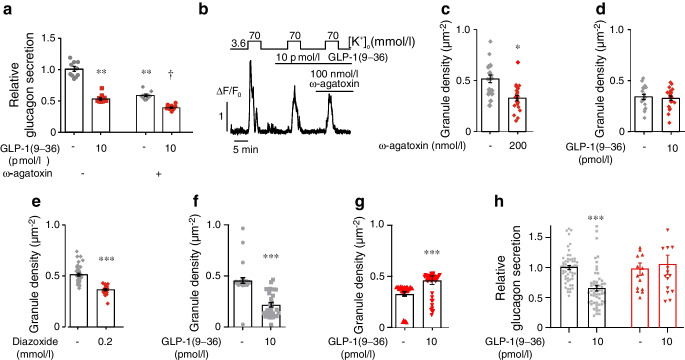
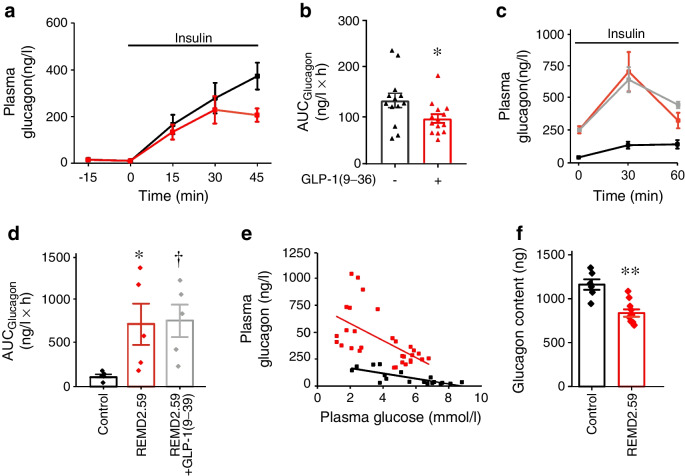
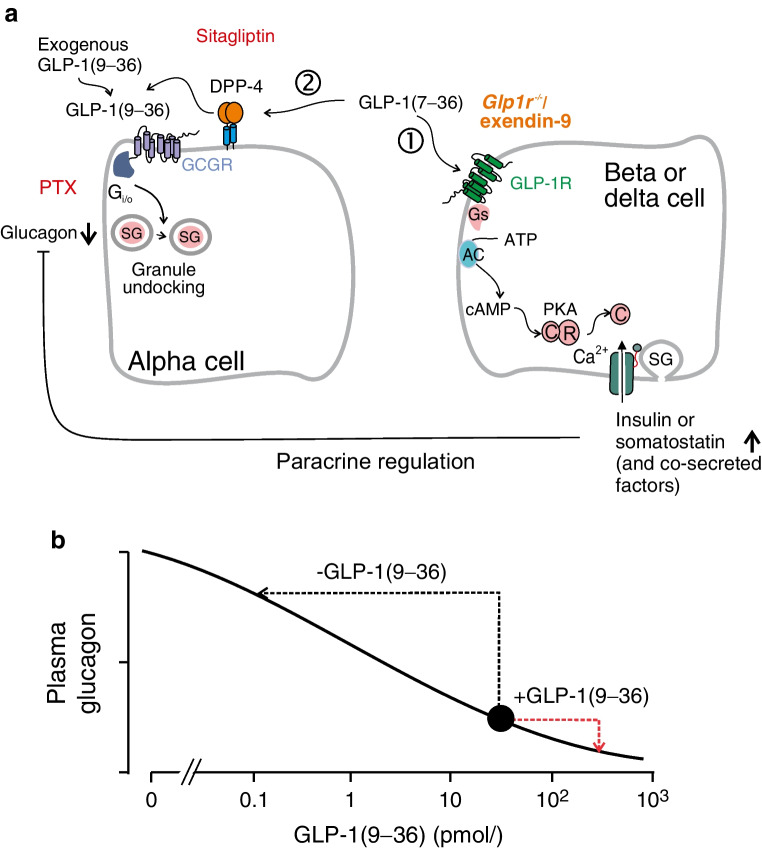
Results: GLP-1(7-36) inhibited glucagon secretion in isolated islets with an IC50 of 2.5 pmol/l. The effect was particularly strong at low glucose concentrations. The degradation product GLP-1(9-36) shared this capacity. GLP-1(9-36) retained its glucagonostatic effects after genetic/pharmacological inactivation of the GLP-1 receptor. GLP-1(9-36) also potently inhibited glucagon secretion evoked by β-adrenergic stimulation, amino acids and membrane depolarisation. In islet alpha cells, GLP-1(9-36) led to inhibition of Ca2+ entry via voltage-gated Ca2+ channels sensitive to ω-agatoxin, with consequential pertussis-toxin-sensitive depletion of the docked pool of secretory granules, effects that were prevented by the glucagon receptor antagonists REMD2.59 and L-168049. The capacity of GLP-1(9-36) to inhibit glucagon secretion and reduce the number of docked granules was lost in alpha cells from human donors with type 2 diabetes. In vivo, high exogenous concentrations of GLP-1(9-36) (>100 pmol/l) resulted in a small (30%) lowering of circulating glucagon during insulin-induced hypoglycaemia. This effect was abolished by REMD2.59, which promptly increased circulating glucagon by >225% (adjusted for the change in plasma glucose) without affecting pancreatic glucagon content.
Conclusions/interpretation: We conclude that the GLP-1 metabolite GLP-1(9-36) is a systemic inhibitor of glucagon secretion. We propose that the increase in circulating glucagon observed following genetic/pharmacological inactivation of glucagon signalling in mice and in people with type 2 diabetes reflects the removal of GLP-1(9-36)'s glucagonostatic action.
Keywords: GLP-1; Glp1r; Glucagon; Glucagon receptor antagonist; Granule docking; Pancreatic alpha cell; Type 2 diabetes.
© 2023. The Author(s).
Figures
Similar articles
-
Glucagon-like peptide-1 and islet lipolysis.Horm Metab Res. 2004 Nov-Dec;36(11-12):795-803. doi: 10.1055/s-2004-826166. Horm Metab Res. 2004. PMID: 15655711
-
Human islets contain a subpopulation of glucagon-like peptide-1 secreting α cells that is increased in type 2 diabetes.Mol Metab. 2020 Sep;39:101014. doi: 10.1016/j.molmet.2020.101014. Epub 2020 May 12. Mol Metab. 2020. PMID: 32413586 Free PMC article.
-
GLP-1 suppresses glucagon secretion in human pancreatic alpha-cells by inhibition of P/Q-type Ca2+ channels.Physiol Rep. 2018 Sep;6(17):e13852. doi: 10.14814/phy2.13852. Physiol Rep. 2018. PMID: 30187652 Free PMC article.
-
The multifaceted potential of glucagon-like peptide-1 as a therapeutic agent.Minerva Endocrinol. 2002 Jun;27(2):79-93. Minerva Endocrinol. 2002. PMID: 11961501 Review.
-
Glucagon-like peptide-1.Recent Prog Horm Res. 2001;56:377-99. doi: 10.1210/rp.56.1.377. Recent Prog Horm Res. 2001. PMID: 11237222 Review.
Cited by
-
Examining the Potential Applicability of Orexigenic and Anorexigenic Peptides in Veterinary Medicine for the Management of Obesity in Companion Animals.Curr Issues Mol Biol. 2024 Jul 1;46(7):6725-6745. doi: 10.3390/cimb46070401. Curr Issues Mol Biol. 2024. PMID: 39057043 Free PMC article. Review.
-
Gut Hormones and Postprandial Metabolic Effects of Isomaltulose vs. Saccharose Consumption in People with Metabolic Syndrome.Nutrients. 2025 Aug 1;17(15):2539. doi: 10.3390/nu17152539. Nutrients. 2025. PMID: 40806123 Free PMC article.
-
Glucagon-like peptide-1 receptor: mechanisms and advances in therapy.Signal Transduct Target Ther. 2024 Sep 18;9(1):234. doi: 10.1038/s41392-024-01931-z. Signal Transduct Target Ther. 2024. PMID: 39289339 Free PMC article. Review.
-
Impact of a 12-Week Dietary Intervention on Adipose Tissue Metabolic Markers in Overweight Women of Reproductive Age.Int J Mol Sci. 2024 Aug 4;25(15):8512. doi: 10.3390/ijms25158512. Int J Mol Sci. 2024. PMID: 39126081 Free PMC article. Clinical Trial.
-
The Importance of Intra-Islet Communication in the Function and Plasticity of the Islets of Langerhans during Health and Diabetes.Int J Mol Sci. 2024 Apr 6;25(7):4070. doi: 10.3390/ijms25074070. Int J Mol Sci. 2024. PMID: 38612880 Free PMC article. Review.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
