

Hyperactive mTORC1 in lung mesenchyme induces endothelial cell dysfunction and pulmonary vascular remodeling
- PMID: 38127441
- PMCID: PMC10866655
- DOI: 10.1172/JCI172116
Hyperactive mTORC1 in lung mesenchyme induces endothelial cell dysfunction and pulmonary vascular remodeling
Abstract
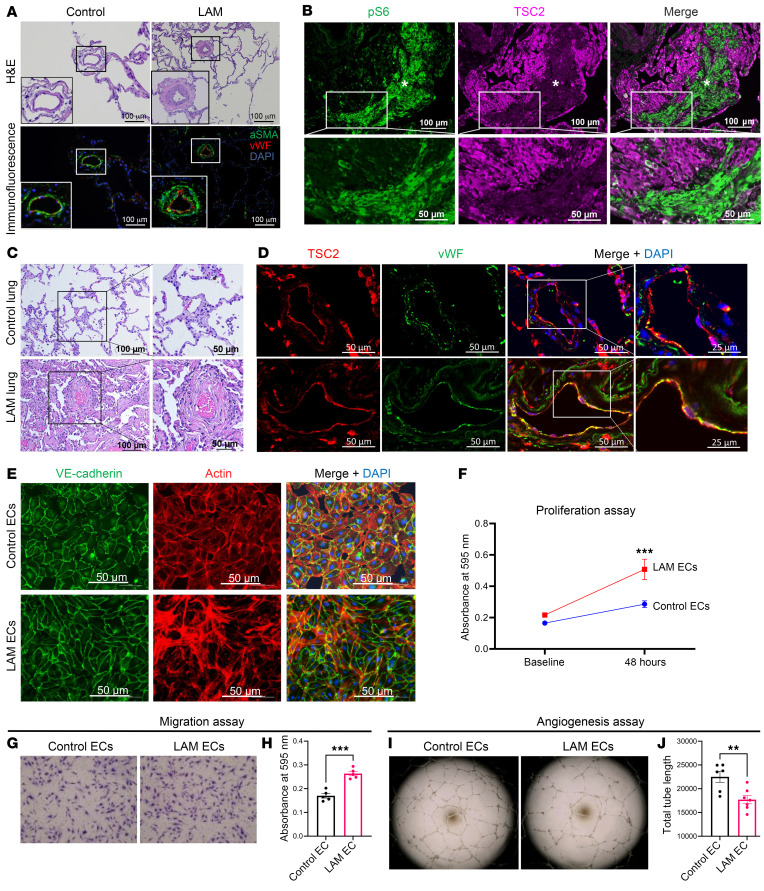
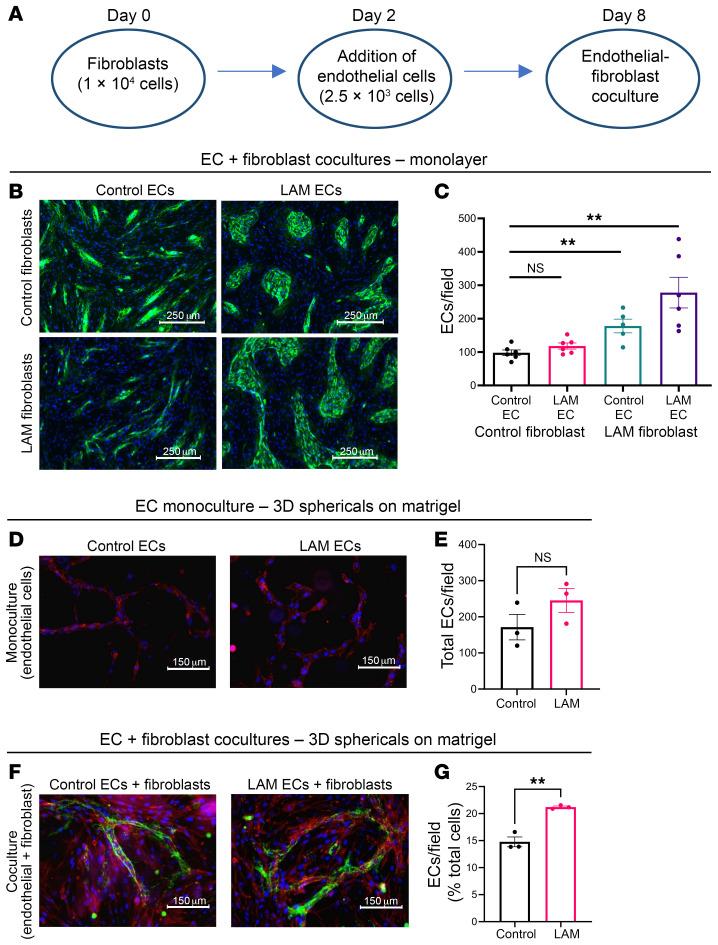
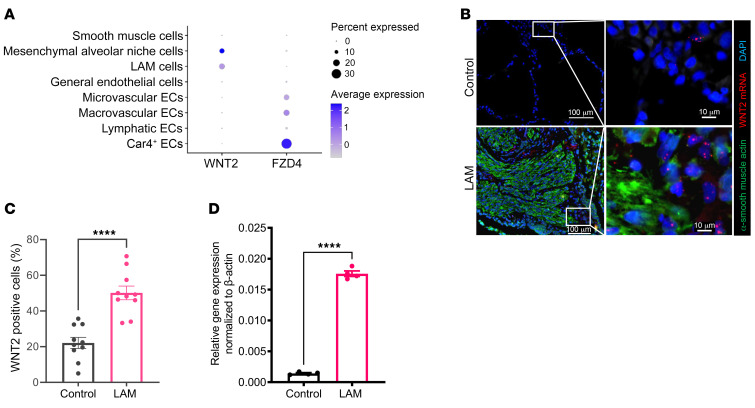
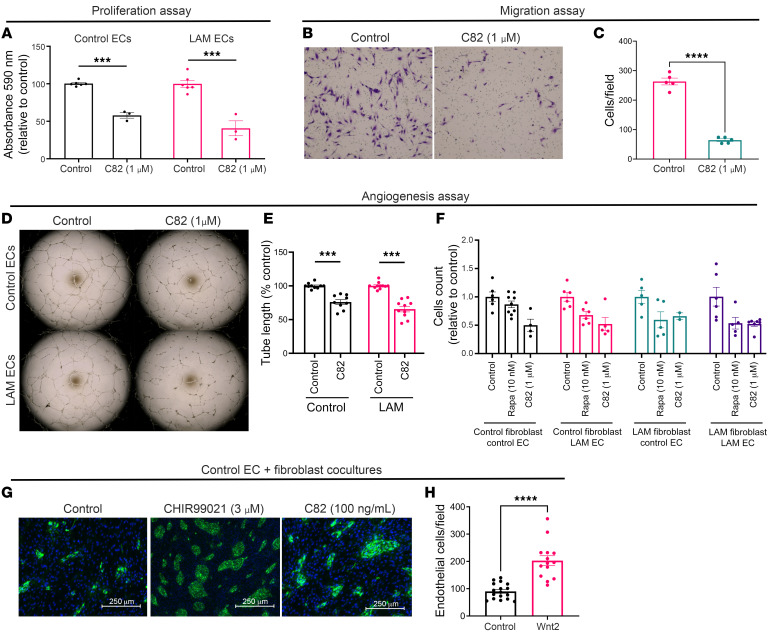
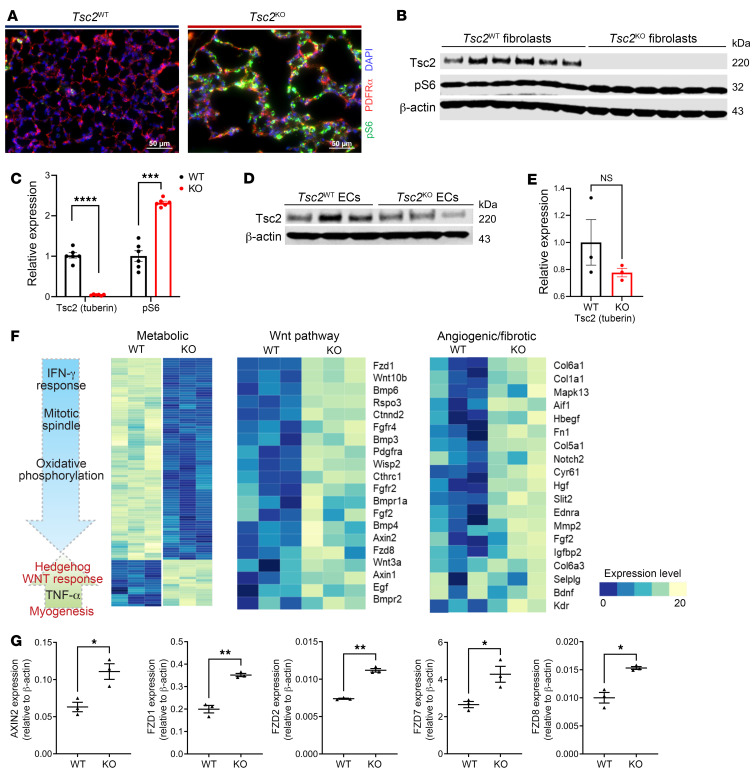
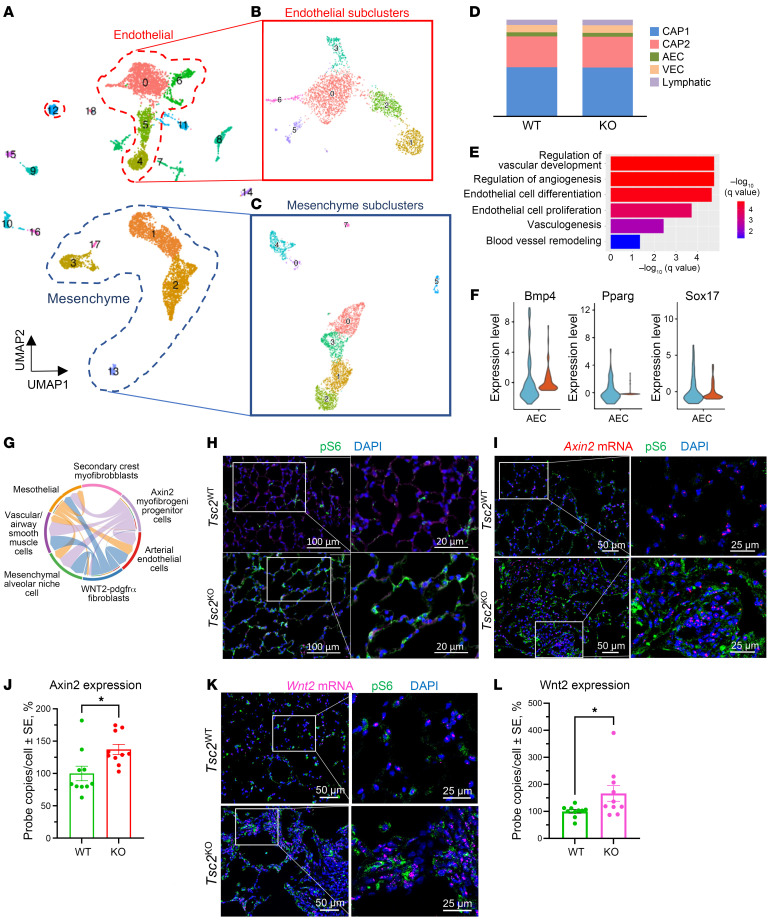
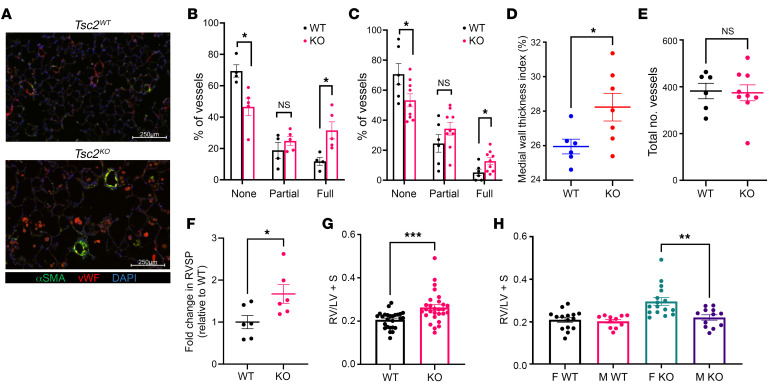
Lymphangioleiomyomatosis (LAM) is a progressive cystic lung disease caused by tuberous sclerosis complex 1/2 (TSC1/2) gene mutations in pulmonary mesenchymal cells, resulting in activation of the mechanistic target of rapamycin complex 1 (mTORC1). A subset of patients with LAM develop pulmonary vascular remodeling and pulmonary hypertension. Little, however, is known regarding how LAM cells communicate with endothelial cells (ECs) to trigger vascular remodeling. In end-stage LAM lung explants, we identified EC dysfunction characterized by increased EC proliferation and migration, defective angiogenesis, and dysmorphic endothelial tube network formation. To model LAM disease, we used an mTORC1 gain-of-function mouse model with a Tsc2 KO (Tsc2KO) specific to lung mesenchyme (Tbx4LME-Cre Tsc2fl/fl), similar to the mesenchyme-specific genetic alterations seen in human disease. As early as 8 weeks of age, ECs from mice exhibited marked transcriptomic changes despite an absence of morphological changes to the distal lung microvasculature. In contrast, 1-year-old Tbx4LME-Cre Tsc2fl/fl mice spontaneously developed pulmonary vascular remodeling with increased medial thickness. Single-cell RNA-Seq of 1-year-old mouse lung cells identified paracrine ligands originating from Tsc2KO mesenchyme, which can signal through receptors in arterial ECs. These ECs had transcriptionally altered genes including those in pathways associated with blood vessel remodeling. The proposed pathophysiologic mesenchymal ligand-EC receptor crosstalk highlights the importance of an altered mesenchymal cell/EC axis in LAM and other hyperactive mTORC1-driven diseases. Since ECs in patients with LAM and in Tbx4LME-Cre Tsc2fl/fl mice did not harbor TSC2 mutations, our study demonstrates that constitutively active mTORC1 lung mesenchymal cells orchestrated dysfunctional EC responses that contributed to pulmonary vascular remodeling.
Keywords: Cell Biology; Endothelial cells; Mouse models; Vascular Biology.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
