

Single-cell characterization of human GBM reveals regional differences in tumor-infiltrating leukocyte activation
- PMID: 38127790
- PMCID: PMC10735226
- DOI: 10.7554/eLife.92678
Single-cell characterization of human GBM reveals regional differences in tumor-infiltrating leukocyte activation
Abstract
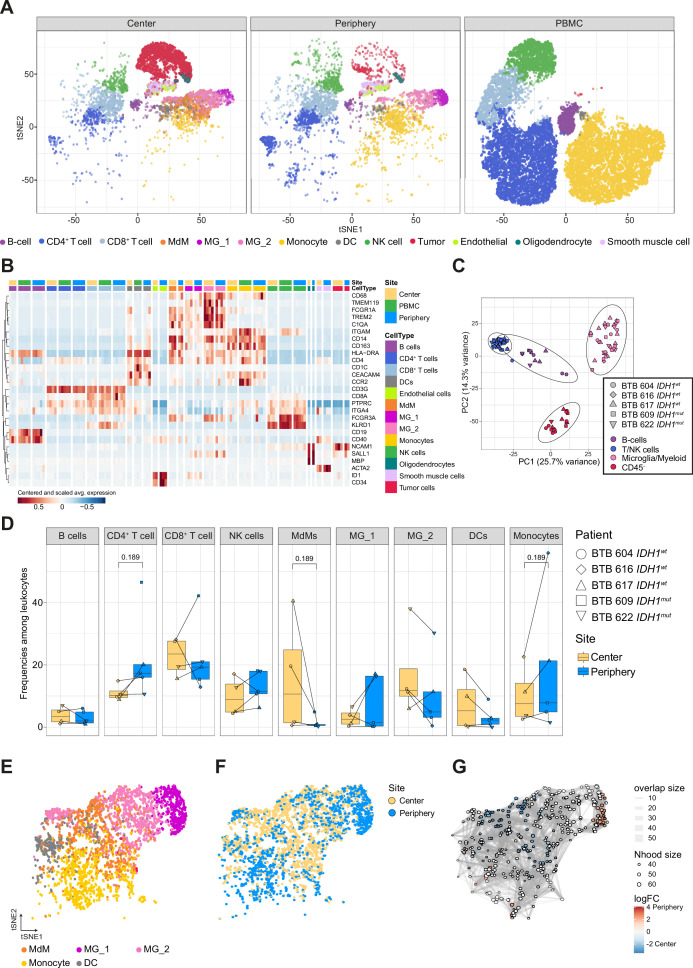
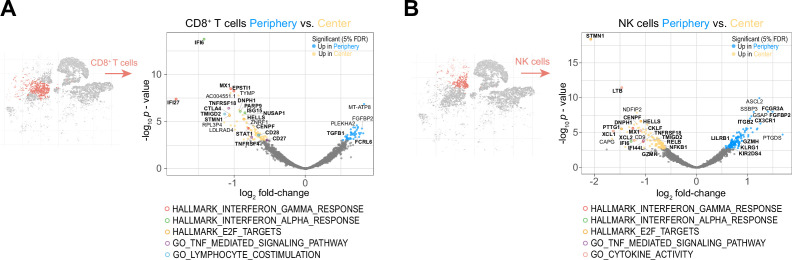
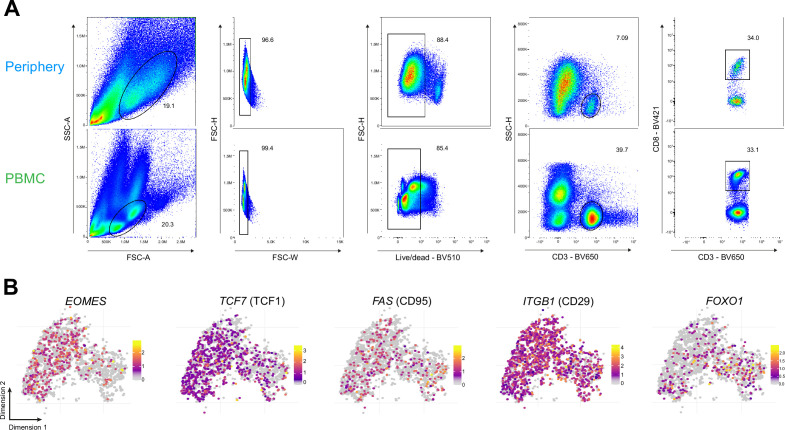
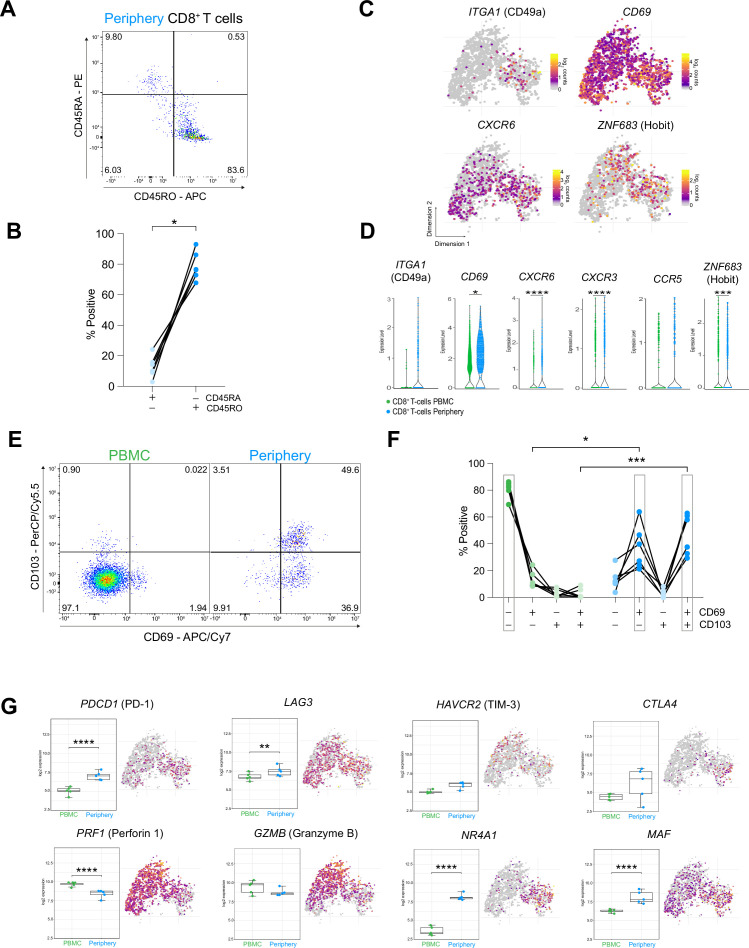
Glioblastoma (GBM) harbors a highly immunosuppressive tumor microenvironment (TME) which influences glioma growth. Major efforts have been undertaken to describe the TME on a single-cell level. However, human data on regional differences within the TME remain scarce. Here, we performed high-depth single-cell RNA sequencing (scRNAseq) on paired biopsies from the tumor center, peripheral infiltration zone and blood of five primary GBM patients. Through analysis of >45,000 cells, we revealed a regionally distinct transcription profile of microglia (MG) and monocyte-derived macrophages (MdMs) and an impaired activation signature in the tumor-peripheral cytotoxic-cell compartment. Comparing tumor-infiltrating CD8+ T cells with circulating cells identified CX3CR1high and CX3CR1int CD8+ T cells with effector and memory phenotype, respectively, enriched in blood but absent in the TME. Tumor CD8+ T cells displayed a tissue-resident memory phenotype with dysfunctional features. Our analysis provides a regionally resolved mapping of transcriptional states in GBM-associated leukocytes, serving as an additional asset in the effort towards novel therapeutic strategies to combat this fatal disease.
Keywords: cancer biology; glioblastoma; human; scRNAseq; tumor microenvironment.
© 2023, Schmassmann et al.
Conflict of interest statement
PS, JR, SH, TS, TM, MR No competing interests declared, SD S.D. is affiliated with Roche Pharmaceutical Research and Early Development. The author has no financial interests to declare, SH S.H. is affiliated with Roche Pharmaceutical Research and Early Development. The author has no financial interests to declare, MB M.B. is affiliated with Roche Pharmaceutical Research and Early Development. The author has no financial interests to declare, GH G.H. has equity in, and is a cofounder of Incephalo Inc
Figures












Update of
References
-
- Abdelfattah N, Kumar P, Wang C, Leu J-S, Flynn WF, Gao R, Baskin DS, Pichumani K, Ijare OB, Wood SL, Powell SZ, Haviland DL, Parker Kerrigan BC, Lang FF, Prabhu SS, Huntoon KM, Jiang W, Kim BYS, George J, Yun K. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nature Communications. 2022;13:767. doi: 10.1038/s41467-022-28372-y. - DOI - PMC - PubMed
-
- Alegre ML, Noel PJ, Eisfelder BJ, Chuang E, Clark MR, Reiner SL, Thompson CB. Regulation of surface and intracellular expression of CTLA4 on mouse T cells. Journal of Immunology. 1996;157:4762–4770. - PubMed
-
- Amezquita RA, Lun ATL, Becht E, Carey VJ, Carpp LN, Geistlinger L, Marini F, Rue-Albrecht K, Risso D, Soneson C, Waldron L, Pagès H, Smith ML, Huber W, Morgan M, Gottardo R, Hicks SC. Orchestrating single-cell analysis with Bioconductor. Nature Methods. 2020;17:137–145. doi: 10.1038/s41592-019-0654-x. - DOI - PMC - PubMed
-
- Aran D, Looney AP, Liu L, Wu E, Fong V, Hsu A, Chak S, Naikawadi RP, Wolters PJ, Abate AR, Butte AJ, Bhattacharya M. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nature Immunology. 2019;20:163–172. doi: 10.1038/s41590-018-0276-y. - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
