

High-throughput PRIME-editing screens identify functional DNA variants in the human genome
- PMID: 38134886
- PMCID: PMC10766087
- DOI: 10.1016/j.molcel.2023.11.021
High-throughput PRIME-editing screens identify functional DNA variants in the human genome
Abstract
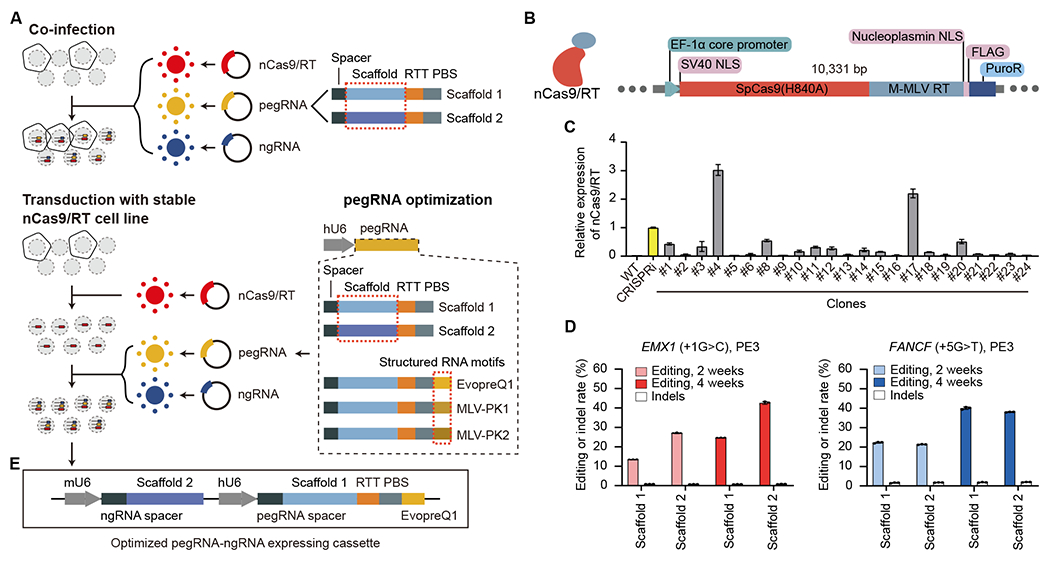
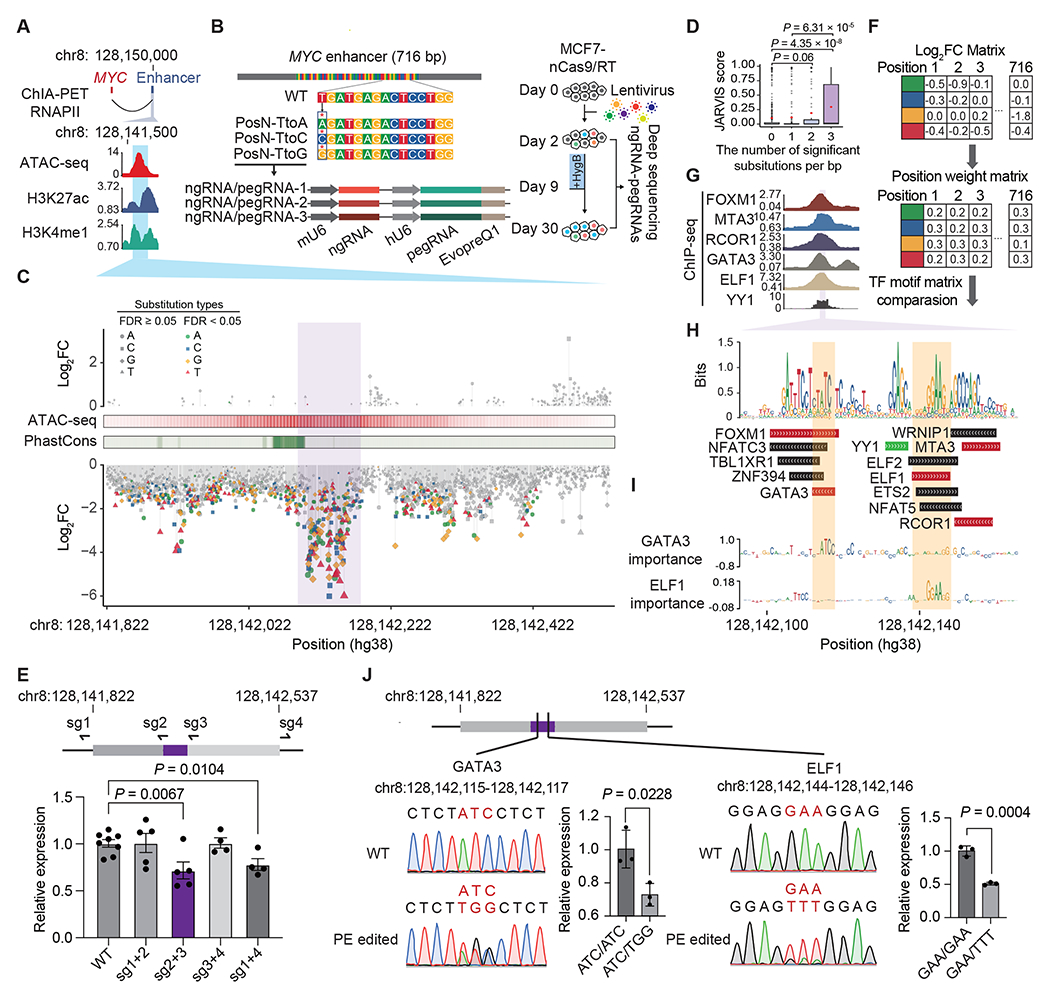
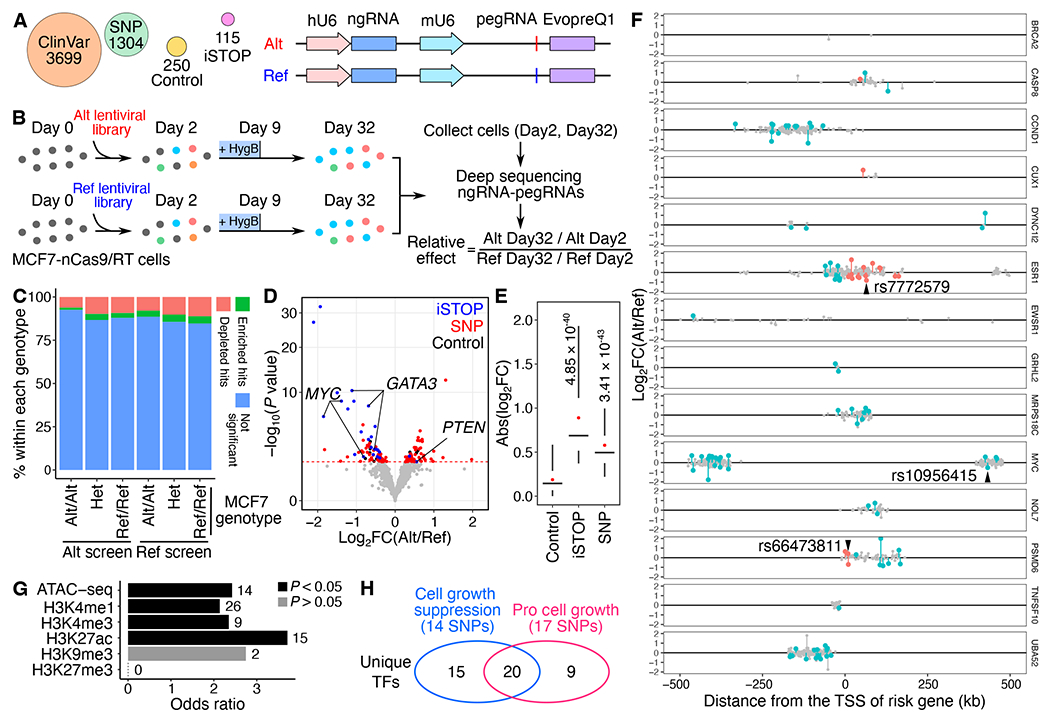
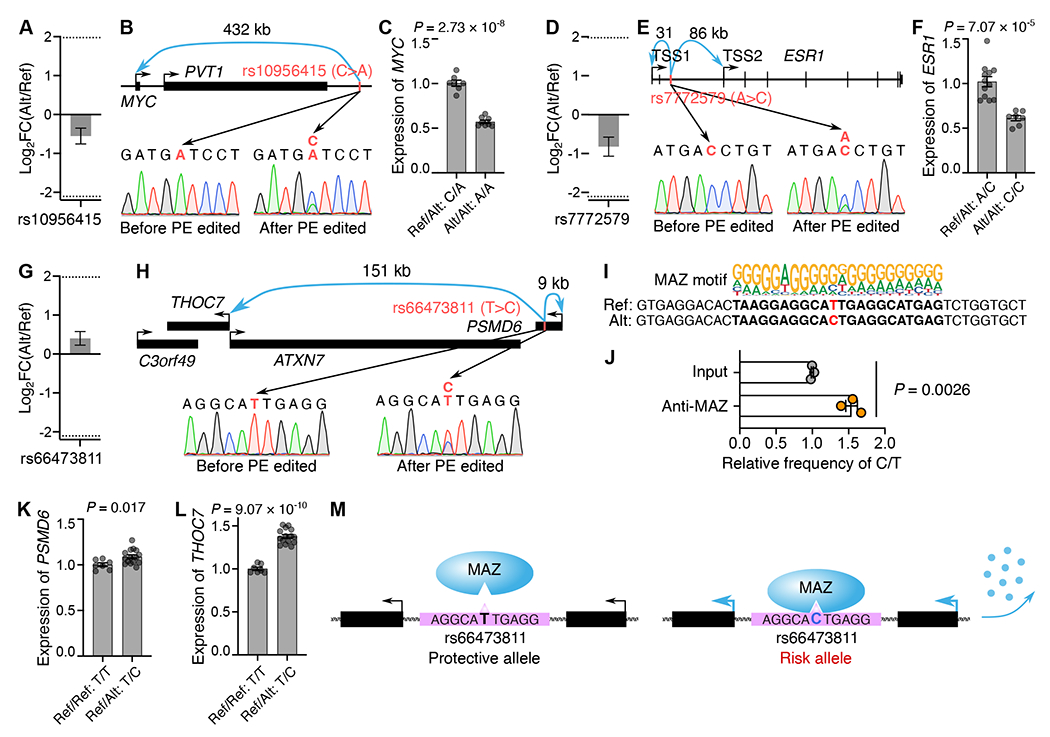
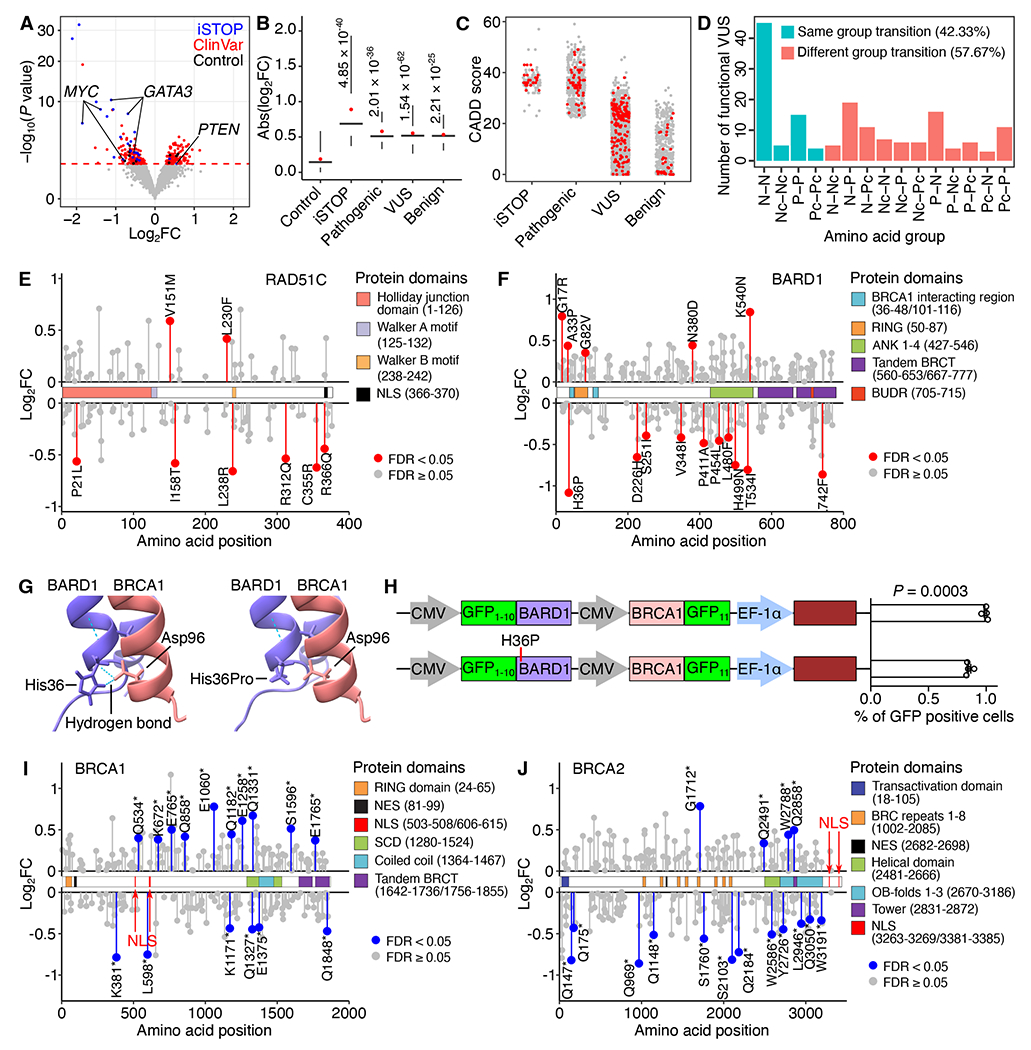
Despite tremendous progress in detecting DNA variants associated with human disease, interpreting their functional impact in a high-throughput and single-base resolution manner remains challenging. Here, we develop a pooled prime-editing screen method, PRIME, that can be applied to characterize thousands of coding and non-coding variants in a single experiment with high reproducibility. To showcase its applications, we first identified essential nucleotides for a 716 bp MYC enhancer via PRIME-mediated single-base resolution analysis. Next, we applied PRIME to functionally characterize 1,304 genome-wide association study (GWAS)-identified non-coding variants associated with breast cancer and 3,699 variants from ClinVar. We discovered that 103 non-coding variants and 156 variants of uncertain significance are functional via affecting cell fitness. Collectively, we demonstrate that PRIME is capable of characterizing genetic variants at single-base resolution and scale, advancing accurate genome annotation for disease risk prediction, diagnosis, and therapeutic target identification.
Keywords: disease variants; enhancer; high-throughput screens; prime editing; single-base resolution.
Copyright © 2023 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests X.R., H.Y., and Yin Shen have filed a patent application related to pooled prime-editing screens.
Figures
Update of
-
High throughput PRIME editing screens identify functional DNA variants in the human genome.bioRxiv [Preprint]. 2023 Jul 12:2023.07.12.548736. doi: 10.1101/2023.07.12.548736. bioRxiv. 2023. Update in: Mol Cell. 2023 Dec 21;83(24):4633-4645.e9. doi: 10.1016/j.molcel.2023.11.021. PMID: 37502948 Free PMC article. Updated. Preprint.
References
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
