

Role of Glycolytic and Glutamine Metabolism Reprogramming on the Proliferation, Invasion, and Apoptosis Resistance through Modulation of Signaling Pathways in Glioblastoma
- PMID: 38139462
- PMCID: PMC10744281
- DOI: 10.3390/ijms242417633
Role of Glycolytic and Glutamine Metabolism Reprogramming on the Proliferation, Invasion, and Apoptosis Resistance through Modulation of Signaling Pathways in Glioblastoma
Abstract
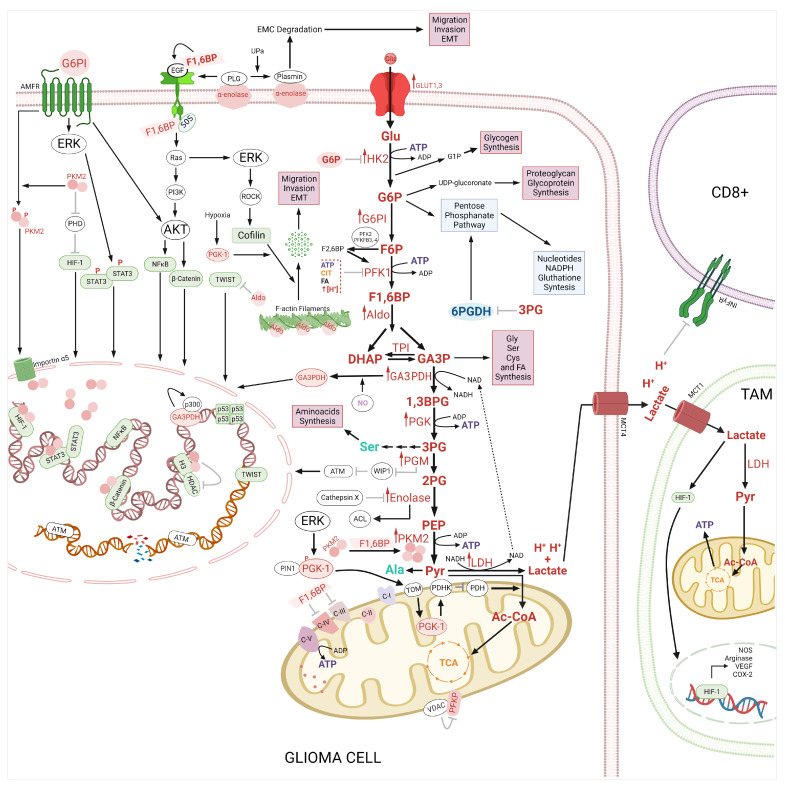
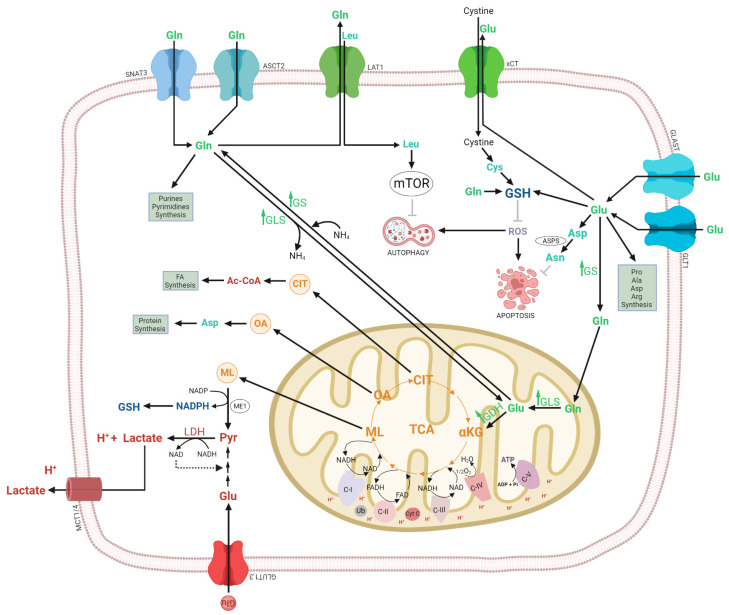
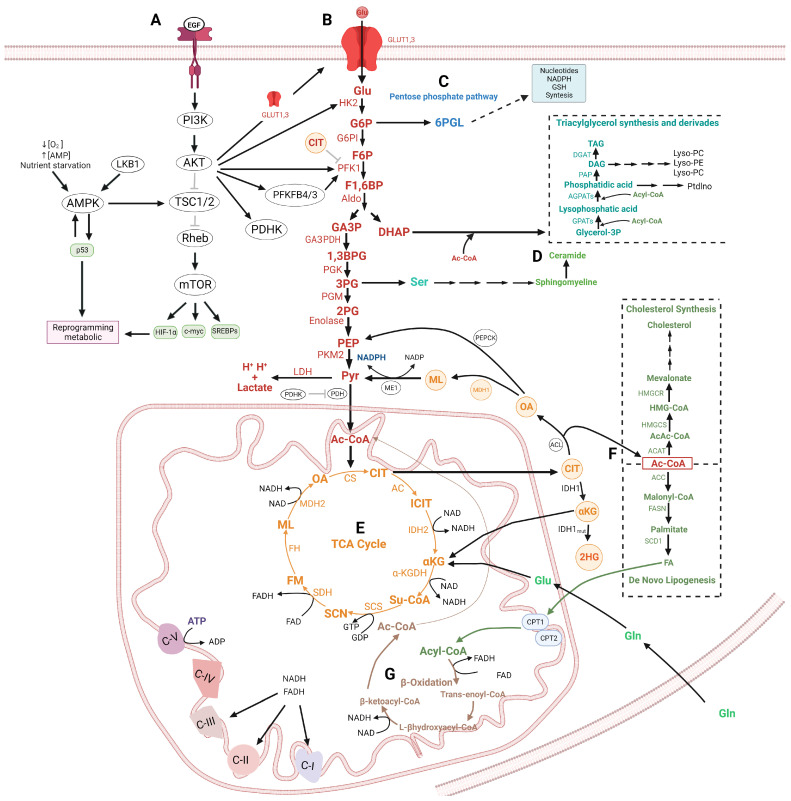
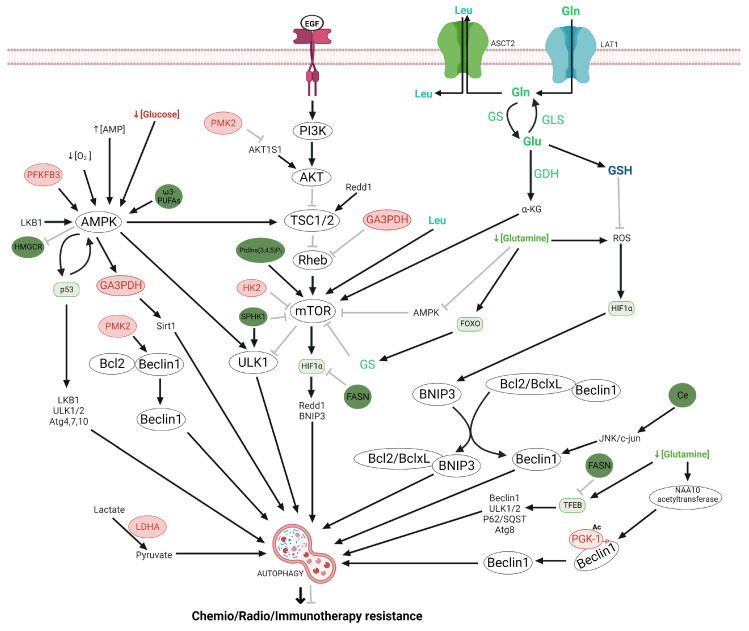
Glioma cells exhibit genetic and metabolic alterations that affect the deregulation of several cellular signal transduction pathways, including those related to glucose metabolism. Moreover, oncogenic signaling pathways induce the expression of metabolic genes, increasing the metabolic enzyme activities and thus the critical biosynthetic pathways to generate nucleotides, amino acids, and fatty acids, which provide energy and metabolic intermediates that are essential to accomplish the biosynthetic needs of glioma cells. In this review, we aim to explore how dysregulated metabolic enzymes and their metabolites from primary metabolism pathways in glioblastoma (GBM) such as glycolysis and glutaminolysis modulate anabolic and catabolic metabolic pathways as well as pro-oncogenic signaling and contribute to the formation, survival, growth, and malignancy of glioma cells. Also, we discuss promising therapeutic strategies by targeting the key players in metabolic regulation. Therefore, the knowledge of metabolic reprogramming is necessary to fully understand the biology of malignant gliomas to improve patient survival significantly.
Keywords: glioma; glucose; glutamine; metabolism; oncogenic pathways.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Metabolic Reprogramming in Glioblastoma Multiforme: A Review of Pathways and Therapeutic Targets.Cells. 2024 Sep 19;13(18):1574. doi: 10.3390/cells13181574. Cells. 2024. PMID: 39329757 Free PMC article. Review.
-
Metabolic dysregulation of tricarboxylic acid cycle and oxidative phosphorylation in glioblastoma.Rev Neurosci. 2024 Jun 7;35(7):813-838. doi: 10.1515/revneuro-2024-0054. Print 2024 Oct 28. Rev Neurosci. 2024. PMID: 38841811 Review.
-
Metabolic reprogramming in cancer cells: glycolysis, glutaminolysis, and Bcl-2 proteins as novel therapeutic targets for cancer.World J Surg Oncol. 2016 Jan 20;14(1):15. doi: 10.1186/s12957-016-0769-9. World J Surg Oncol. 2016. PMID: 26791262 Free PMC article. Review.
-
Glutamine Addiction In Gliomas.Neurochem Res. 2017 Jun;42(6):1735-1746. doi: 10.1007/s11064-017-2212-1. Epub 2017 Mar 9. Neurochem Res. 2017. PMID: 28281102 Review.
-
Clinical research framework proposal for ketogenic metabolic therapy in glioblastoma.BMC Med. 2024 Dec 5;22(1):578. doi: 10.1186/s12916-024-03775-4. BMC Med. 2024. PMID: 39639257 Free PMC article. Review.
Cited by
-
Gut microbiota's role in glioblastoma risk, with a focus on the mediating role of metabolites.Front Neurol. 2024 Jul 3;15:1386885. doi: 10.3389/fneur.2024.1386885. eCollection 2024. Front Neurol. 2024. PMID: 39022732 Free PMC article.
-
Metabolic Reprogramming in Glioblastoma Multiforme: A Review of Pathways and Therapeutic Targets.Cells. 2024 Sep 19;13(18):1574. doi: 10.3390/cells13181574. Cells. 2024. PMID: 39329757 Free PMC article. Review.
-
Identification of Prognostic Genes Related to Cell Senescence and Lipid Metabolism in Glioblastoma Based on Transcriptome and Single-Cell RNA-Seq Data.Int J Mol Sci. 2025 Feb 21;26(5):1875. doi: 10.3390/ijms26051875. Int J Mol Sci. 2025. PMID: 40076502 Free PMC article.
-
Nontargeted metabolomics uncovering metabolite signatures in glioblastoma: a preliminary study on candidate biomarker discovery for IDH subtyping and survival prediction.Front Oncol. 2025 May 8;15:1568040. doi: 10.3389/fonc.2025.1568040. eCollection 2025. Front Oncol. 2025. PMID: 40406272 Free PMC article.
-
Metabolic flux analysis of glioblastoma neural stem cells reveals distinctive metabolic phenotypes in ketogenic conditions.Sci Rep. 2025 May 28;15(1):18736. doi: 10.1038/s41598-025-02124-6. Sci Rep. 2025. PMID: 40436928 Free PMC article.
References
-
- Louis D.N., Perry A., Reifenberger G., von Deimling A., Figarella-Branger D., Cavenee W.K., Ohgaki H., Wiestler O.D., Kleihues P., Ellison D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
