

Targeting the tissue factor coagulation initiation complex prevents antiphospholipid antibody development
- PMID: 38142429
- PMCID: PMC10972716
- DOI: 10.1182/blood.2023022276
Targeting the tissue factor coagulation initiation complex prevents antiphospholipid antibody development
Abstract
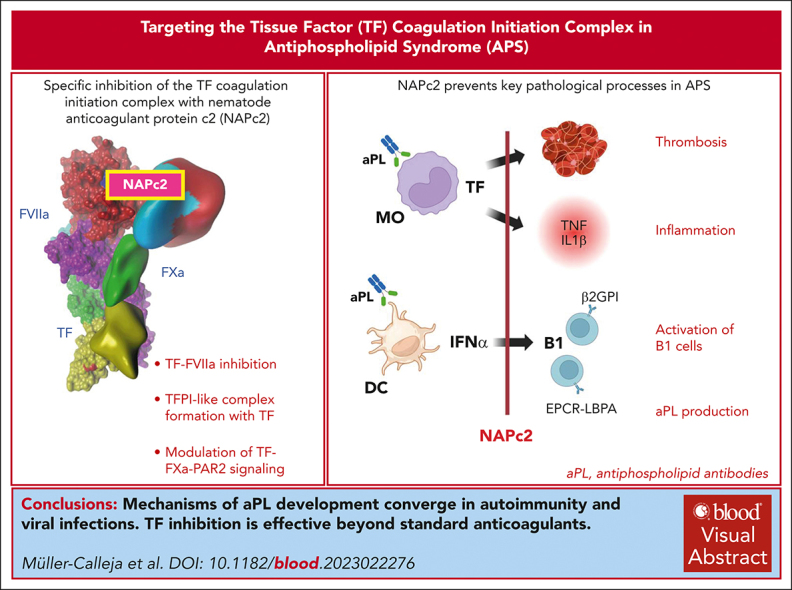
Antiphospholipid antibodies (aPL) in primary or secondary antiphospholipid syndrome (APS) are a major cause for acquired thrombophilia, but specific interventions preventing autoimmune aPL development are an unmet clinical need. Although autoimmune aPL cross react with various coagulation regulatory proteins, lipid-reactive aPL, including those derived from patients with COVID-19, recognize the endolysosomal phospholipid lysobisphosphatidic acid presented by the cell surface-expressed endothelial protein C receptor. This specific recognition leads to complement-mediated activation of tissue factor (TF)-dependent proinflammatory signaling and thrombosis. Here, we show that specific inhibition of the TF coagulation initiation complex with nematode anticoagulant protein c2 (NAPc2) prevents the prothrombotic effects of aPL derived from patients with COVID-19 in mice and the aPL-induced proinflammatory and prothrombotic activation of monocytes. The induction of experimental APS is dependent on the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, and NAPc2 suppresses monocyte endosomal reactive oxygen species production requiring the TF cytoplasmic domain and interferon-α secretion from dendritic cells. Latent infection with murine cytomegalovirus causes TF cytoplasmic domain-dependent development of persistent aPL and circulating phospholipid-reactive B1 cells, which is prevented by short-term intervention with NAPc2 during acute viral infection. In addition, treatment of lupus prone MRL-lpr mice with NAPc2, but not with heparin, suppresses dendritic-cell activation in the spleen, aPL production and circulating phospholipid-reactive B1 cells, and attenuates lupus pathology. These data demonstrate a convergent TF-dependent mechanism of aPL development in latent viral infection and autoimmune disease and provide initial evidence that specific targeting of the TF initiation complex has therapeutic benefits beyond currently used clinical anticoagulant strategies.
© 2024 American Society of Hematology. Published by Elsevier Inc. All rights are reserved, including those for text and data mining, AI training, and similar technologies.
Conflict of interest statement
Conflict-of-interest disclosure: W.R. is a consultant for ARCA Biopharma. W.R., N.M.-C., K.L., and J.W.-M. are listed as inventors on intellectual properties for the therapeutic use of NAPc2. The remaining authors declare no competing financial interests.
Figures







Comment in
-
The mystery of autoantibodies solved?Blood. 2024 Mar 21;143(12):1065-1066. doi: 10.1182/blood.2023023637. Blood. 2024. PMID: 38512263 No abstract available.
References
-
- Garcia D, Erkan D. Diagnosis and management of the antiphospholipid syndrome. N Engl J Med. 2018;378(21):2010–2021. - PubMed
-
- Schreiber K, Sciascia S, de Groot PG, et al. Antiphospholipid syndrome. Nat Rev Dis Primers. 2018;4 - PubMed
-
- Knight JS, Branch DW, Ortel TL. Antiphospholipid syndrome: advances in diagnosis, pathogenesis, and management. BMJ. 2023;380 - PubMed
-
- Passam FH, Chen G, Chen VM, Qi M, Krilis SA, Giannakopoulos B. Betaeta-2-glycoprotein I exerts antithrombotic function through its domain V in mice. J Autoimmun. 2022;126 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
