

Druggable genomic landscapes of high-grade gliomas
- PMID: 38143440
- PMCID: PMC10749203
- DOI: 10.3389/fmed.2023.1254955
Druggable genomic landscapes of high-grade gliomas
Abstract
Background: Despite the putatively targetable genomic landscape of high-grade gliomas, the long-term survival benefit of genomically-tailored targeted therapies remains discouraging.
Methods: Using glioblastoma (GBM) as a representative example of high-grade gliomas, we evaluated the clonal architecture and distribution of hotspot mutations in 388 GBMs from the Cancer Genome Atlas (TCGA). Mutations were matched with 54 targeted therapies, followed by a comprehensive evaluation of drug biochemical properties in reference to the drug's clinical efficacy in high-grade gliomas. We then assessed clinical outcomes of a cohort of patients with high-grade gliomas with targetable mutations reviewed at the Johns Hopkins Molecular Tumor Board (JH MTB; n = 50).
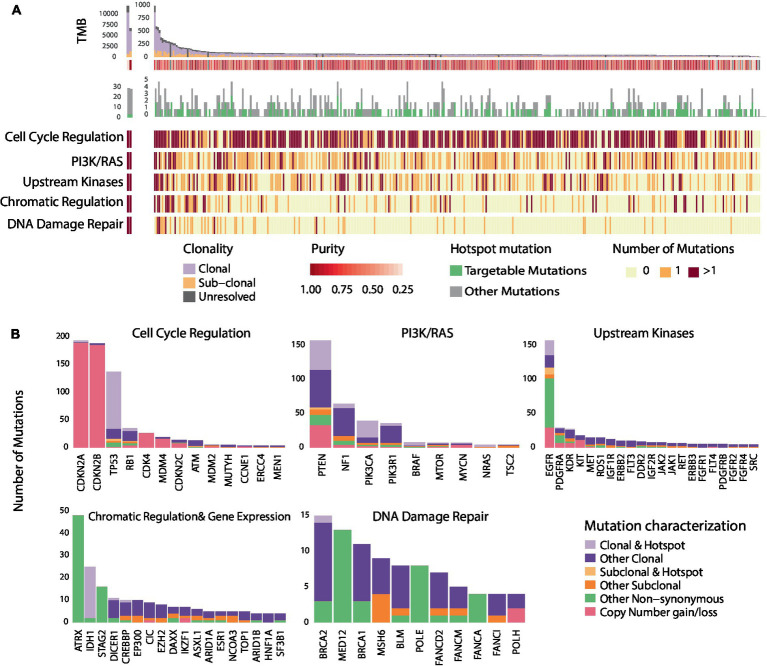
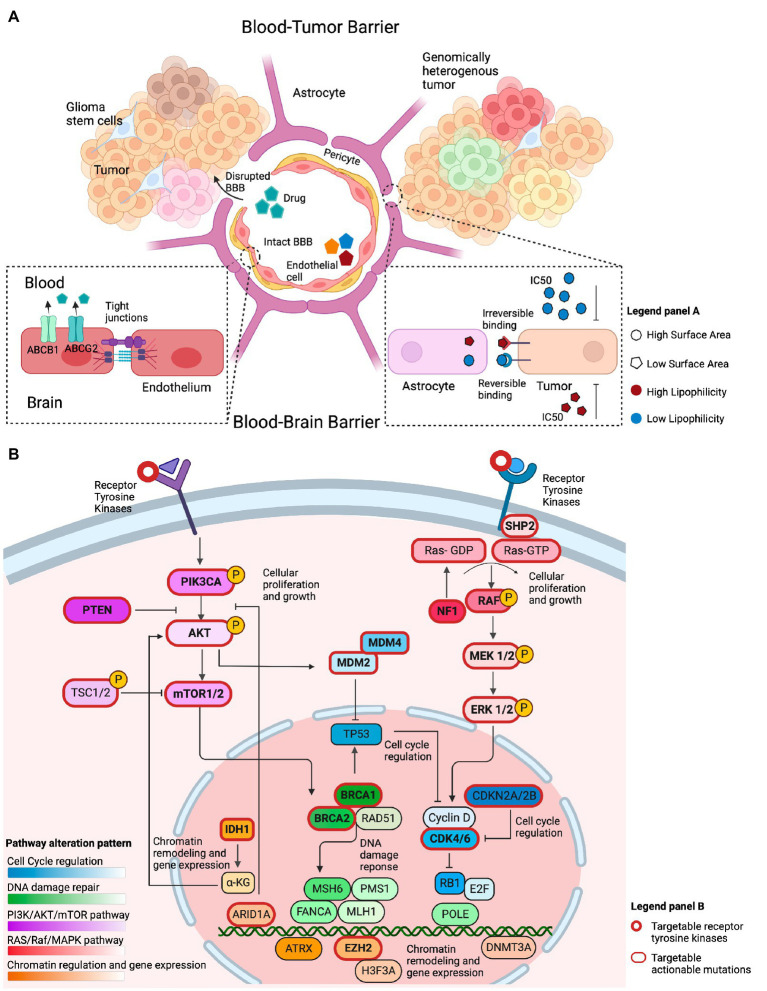
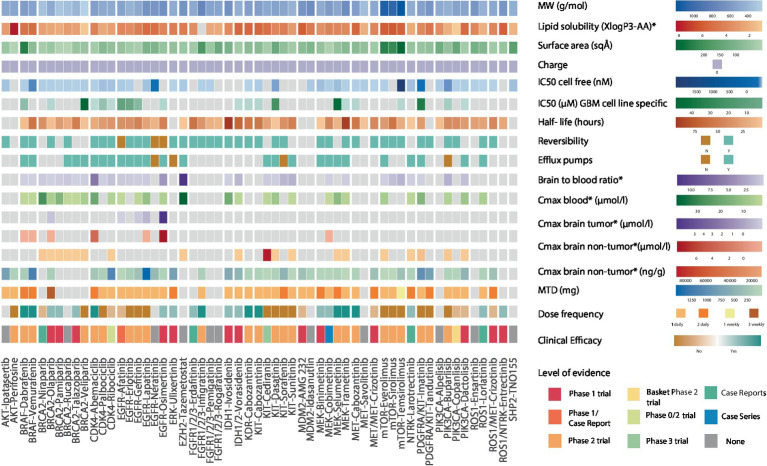
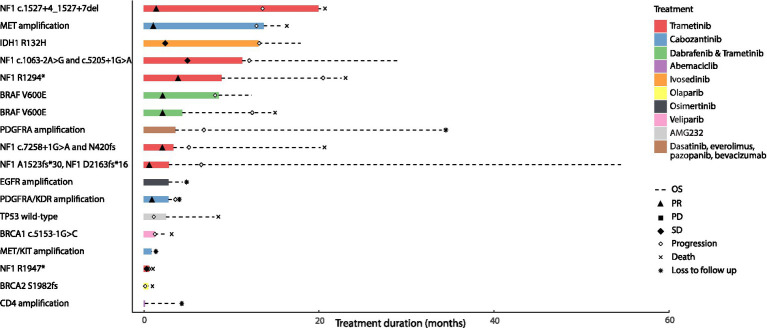
Results: Among 1,156 sequence alterations evaluated, 28.6% represented hotspots. While the frequency of hotspot mutations in GBM was comparable to cancer types with actionable hotspot alterations, GBMs harbored a higher fraction of subclonal mutations that affected hotspots (7.0%), compared to breast cancer (4.9%), lung cancer (4.4%), and melanoma (1.4%). In investigating the biochemical features of targeted therapies paired with recurring alterations, we identified a trend toward higher lipid solubility and lower IC50 in GBM cell lines among drugs with clinical efficacy. The drugs' half-life, molecular weight, surface area and binding to efflux transporters were not associated with clinical efficacy. Among the JH MTB cohort of patients with IDH1 wild-type high-grade gliomas who received targeted therapies, trametinib monotherapy or in combination with dabrafenib conferred radiographic partial response in 75% of patients harboring BRAF or NF1 actionable mutations. Cabozantinib conferred radiographic partial response in two patients harboring a MET and a PDGFRA/KDR amplification. Patients with IDH1 wild-type gliomas that harbored actionable alterations who received genotype-matched targeted therapy had longer progression-free (PFS) and overall survival (OS; 7.37 and 14.72 respectively) than patients whose actionable alterations were not targeted (2.83 and 4.2 months respectively).
Conclusion: While multiple host, tumor and drug-related features may limit the delivery and efficacy of targeted therapies for patients with high-grade gliomas, genotype-matched targeted therapies confer favorable clinical outcomes. Further studies are needed to generate more data on the impact of biochemical features of targeted therapies on their clinical efficacy for high-grade gliomas.
Keywords: actionable mutation; genomic landscape; glioblastoma; glioma; molecular tumor board; precision oncology; targeted therapies.
Copyright © 2023 Ghanem, Fatteh, Kamson, Balan, Chang, Tao, Blakeley, the Johns Hopkins Molecular Tumor Board Investigators, Canzoniero, Grossman, Marrone, Schreck and Anagnostou.
Conflict of interest statement
VA receives research funding to Johns Hopkins University from Astra Zeneca and Personal Genome Diagnostics, has received research funding to Johns Hopkins University from Bristol-Myers Squibb and Delfi Diagnostics in the past 5 years, is an advisory board member for Neogenomics and receives honoraria from Foundation Medicine. KM is a consultant for Astra Zeneca, Amgen, Puma Biotechnology, Jannsen, Mirati Therapeutics, Daiichi Sankyo/Lilly and Regeneron, receives research funding from Mirati, Bristol-Myers Squibb and Astra Zeneca. JC is a consultant for Illumina. KS serves on a data safety and monitoring committee for Advarra, has received honoraria from Springworks Therapeutics and receives research funding to Johns Hopkins University from Springworks Therapeutics. JB serves on an advisory board for Springworks Therapeutics. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJB, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7, PMID: - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous