

Collaborative effect of Csnk1a1 haploinsufficiency and mutant p53 in Myc induction can promote leukemic transformation
- PMID: 38147624
- PMCID: PMC10847877
- DOI: 10.1182/bloodadvances.2022008926
Collaborative effect of Csnk1a1 haploinsufficiency and mutant p53 in Myc induction can promote leukemic transformation
Abstract
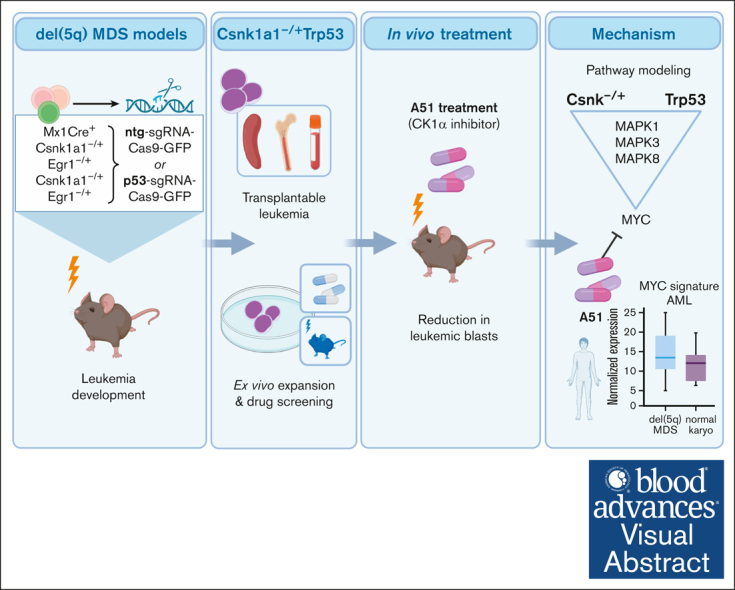
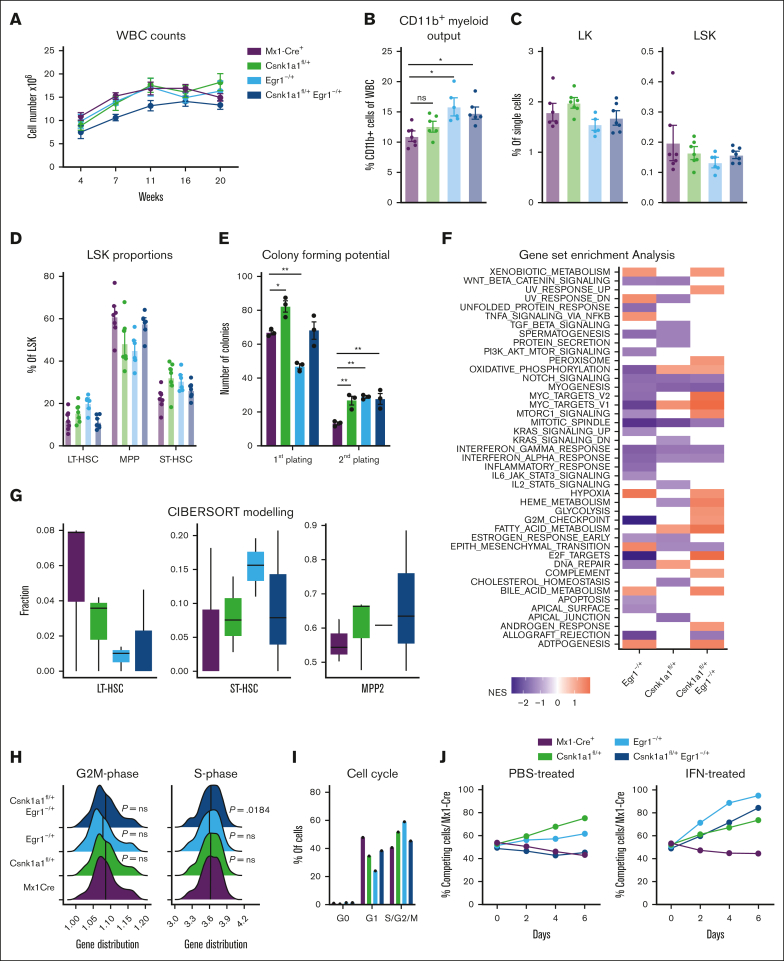
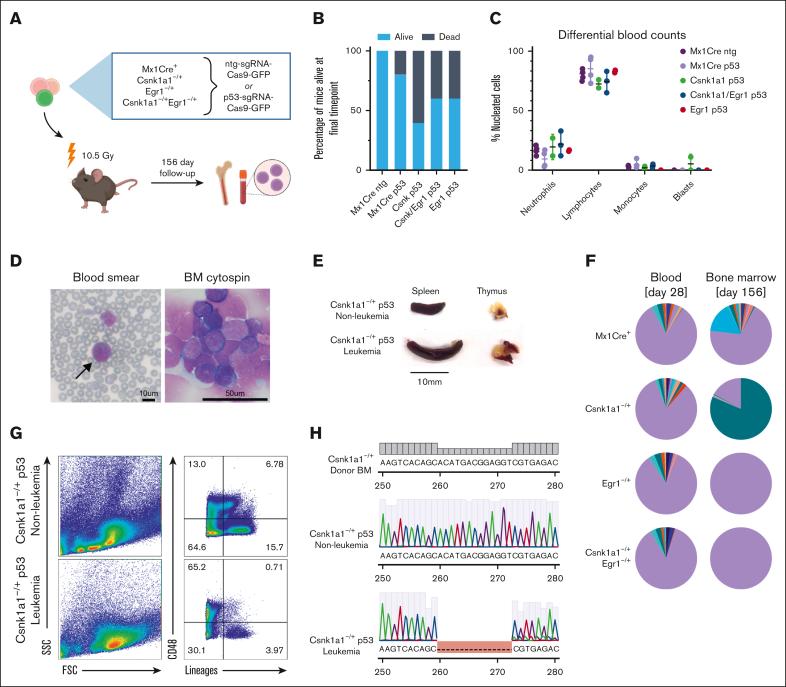
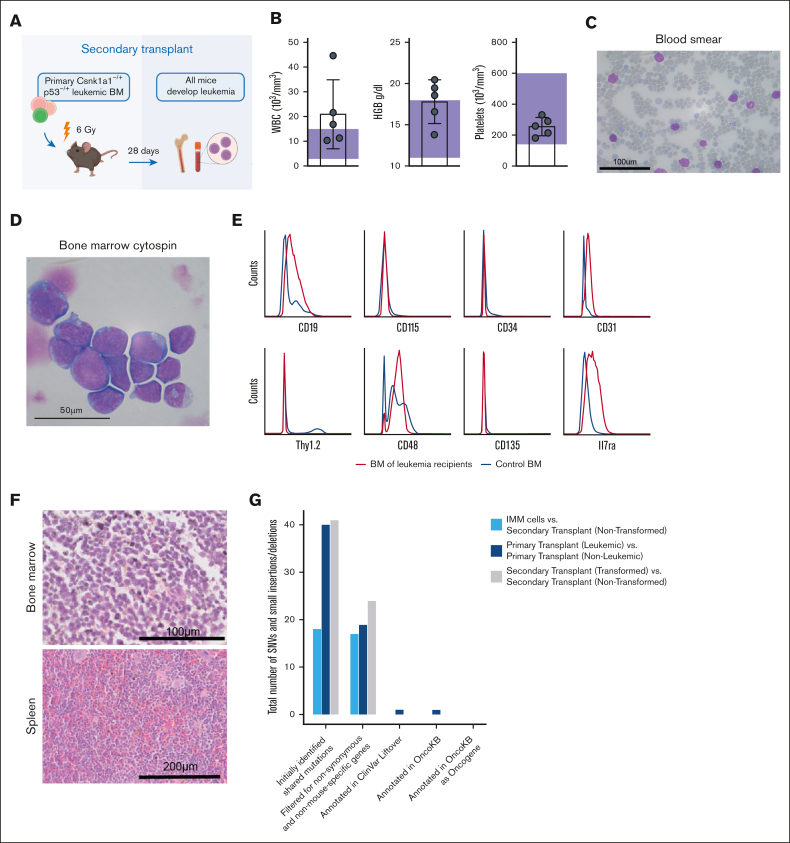
It is still not fully understood how genetic haploinsufficiency in del(5q) myelodysplastic syndrome (MDS) contributes to malignant transformation of hematopoietic stem cells. We asked how compound haploinsufficiency for Csnk1a1 and Egr1 in the common deleted region on chromosome 5 affects hematopoietic stem cells. Additionally, Trp53 was disrupted as the most frequently comutated gene in del(5q) MDS using CRISPR/Cas9 editing in hematopoietic progenitors of wild-type (WT), Csnk1a1-/+, Egr1-/+, Csnk1a1/Egr1-/+ mice. A transplantable acute leukemia only developed in the Csnk1a1-/+Trp53-edited recipient. Isolated blasts were indefinitely cultured ex vivo and gave rise to leukemia after transplantation, providing a tool to study disease mechanisms or perform drug screenings. In a small-scale drug screening, the collaborative effect of Csnk1a1 haploinsufficiency and Trp53 sensitized blasts to the CSNK1 inhibitor A51 relative to WT or Csnk1a1 haploinsufficient cells. In vivo, A51 treatment significantly reduced blast counts in Csnk1a1 haploinsufficient/Trp53 acute leukemias and restored hematopoiesis in the bone marrow. Transcriptomics on blasts and their normal counterparts showed that the derived leukemia was driven by MAPK and Myc upregulation downstream of Csnk1a1 haploinsufficiency cooperating with a downregulated p53 axis. A collaborative effect of Csnk1a1 haploinsufficiency and p53 loss on MAPK and Myc upregulation was confirmed on the protein level. Downregulation of Myc protein expression correlated with efficient elimination of blasts in A51 treatment. The "Myc signature" closely resembled the transcriptional profile of patients with del(5q) MDS with TP53 mutation.
© 2024 by The American Society of Hematology. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: Y.B.-N. is a consultant for Edgewood Oncology, a company advancing A51 clinical development for AML/high-risk MDS. The remaining authors declare no competing financial interests.
Figures
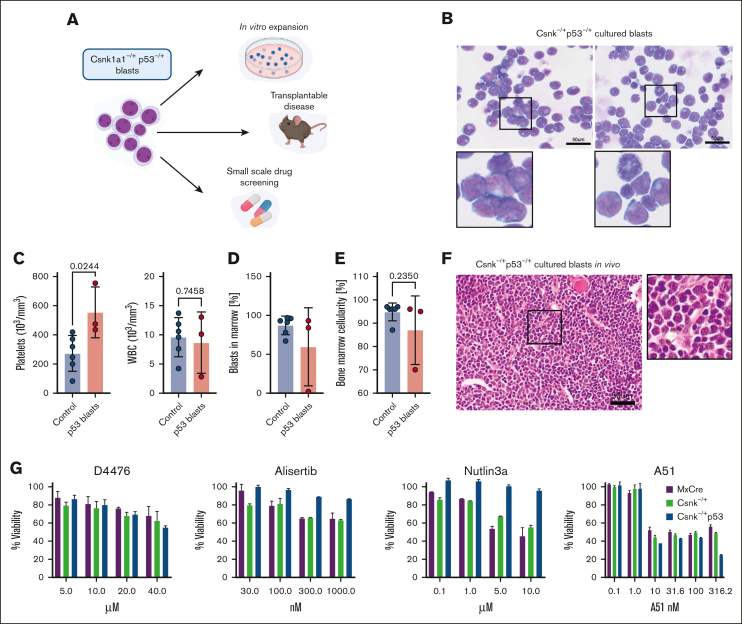

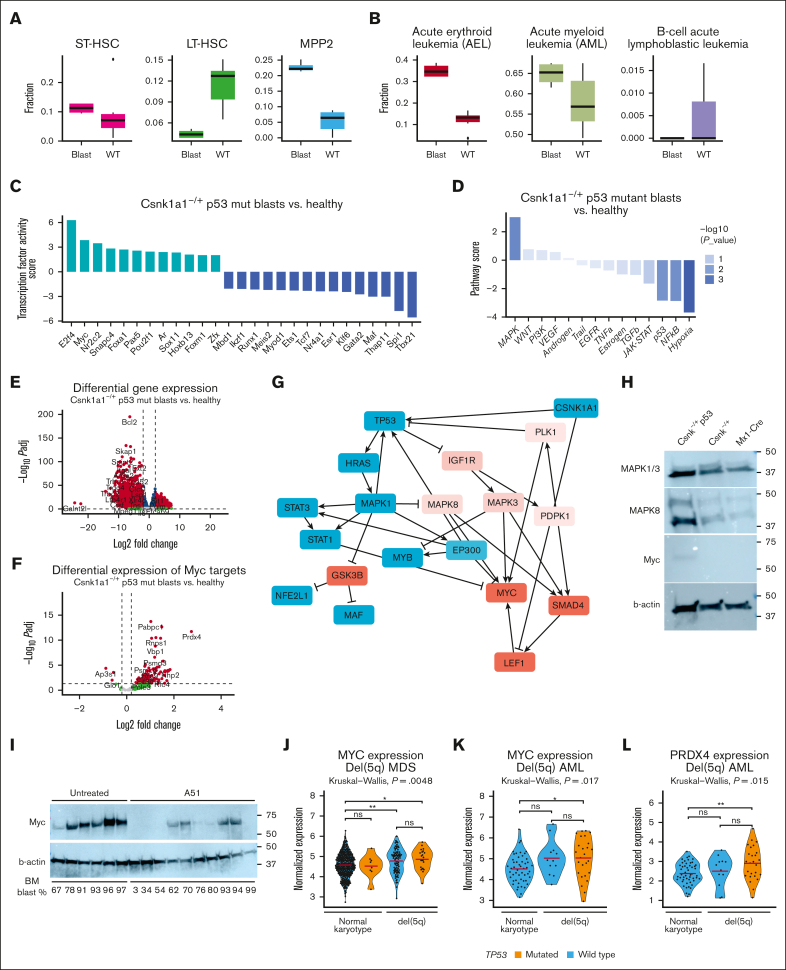
References
-
- Haase D, Germing U, Schanz J, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110(13):4385–4395. - PubMed
-
- Christiansen DH, Andersen MK, Pedersen-Bjergaard J. Mutations with loss of heterozygosity of p53 are common in therapy-related myelodysplasia and acute myeloid leukemia after exposure to alkylating agents and significantly associated with deletion or loss of 5q, a complex karyotype, and a poor prognosis. J Clin Oncol. 2001;19(5):1405–1413. - PubMed
-
- Rücker FG, Schlenk RF, Bullinger L, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119(9):2114–2121. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
