

Molecular characterization of chronic cutaneous wounds reveals subregion- and wound type-specific differential gene expression
- PMID: 38149752
- PMCID: PMC10958103
- DOI: 10.1111/iwj.14447
Molecular characterization of chronic cutaneous wounds reveals subregion- and wound type-specific differential gene expression
Abstract
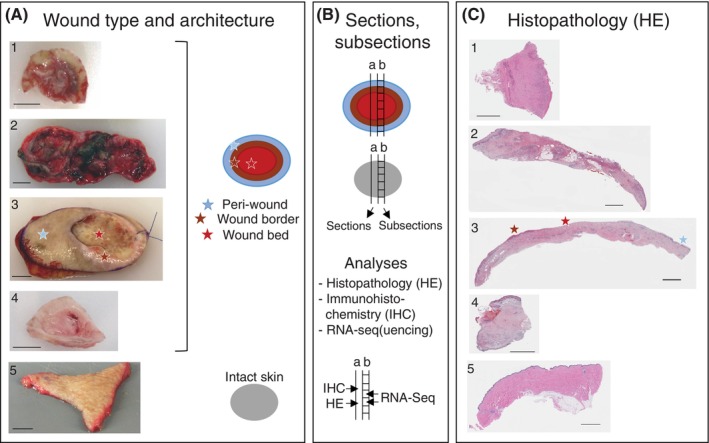
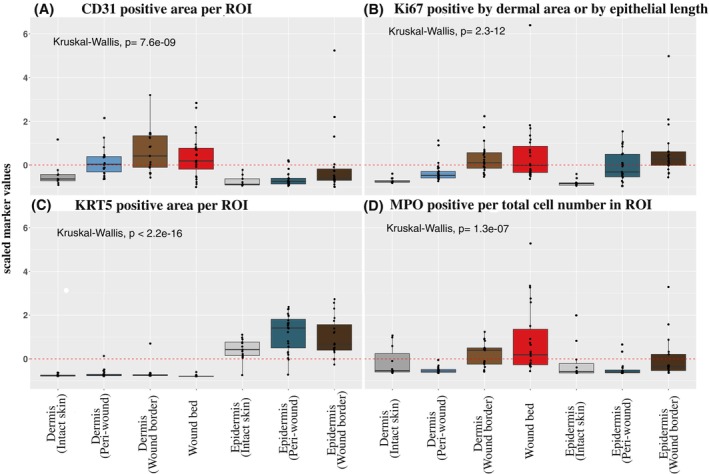
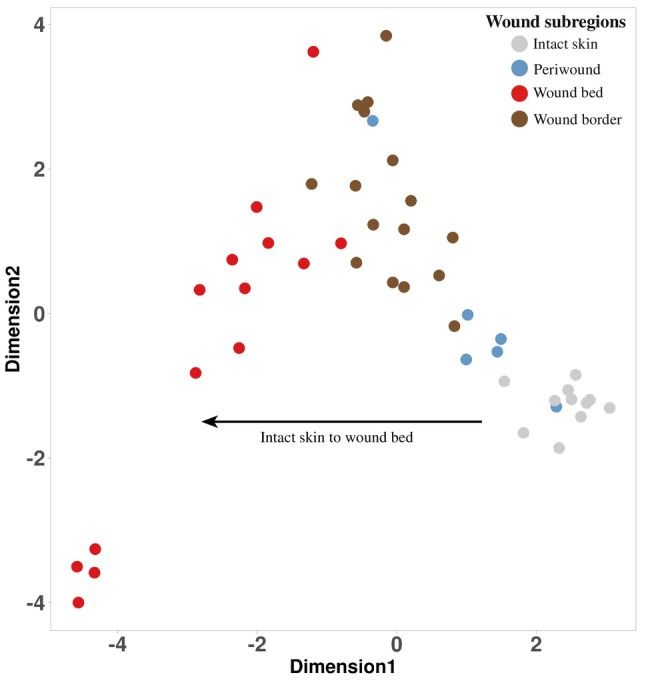
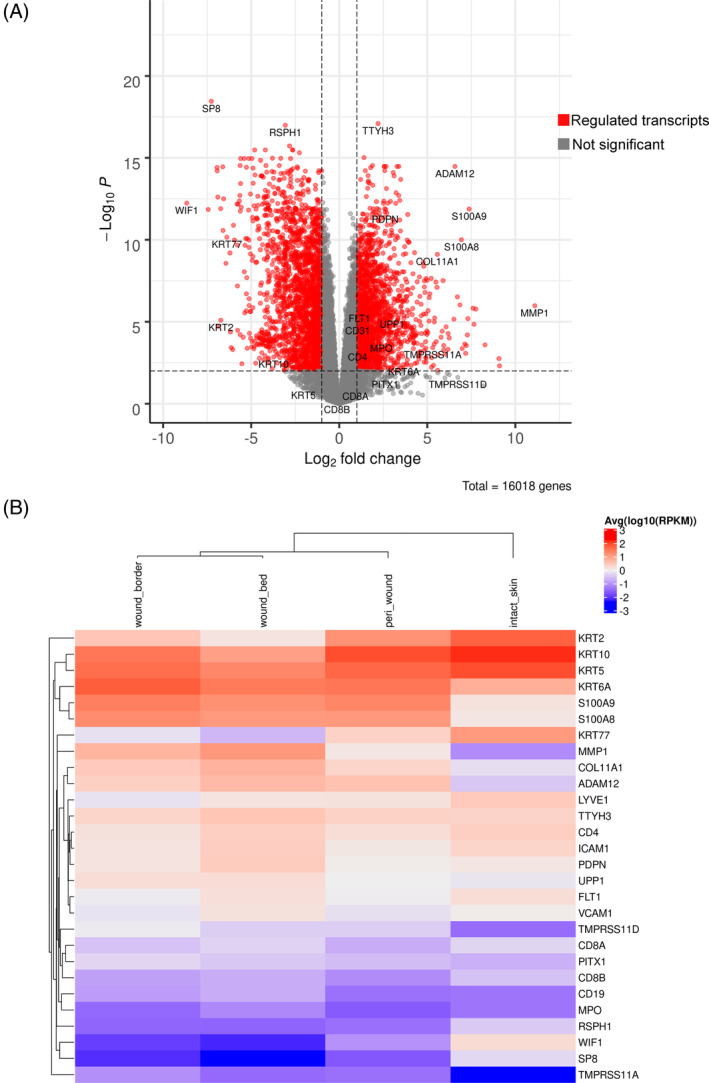
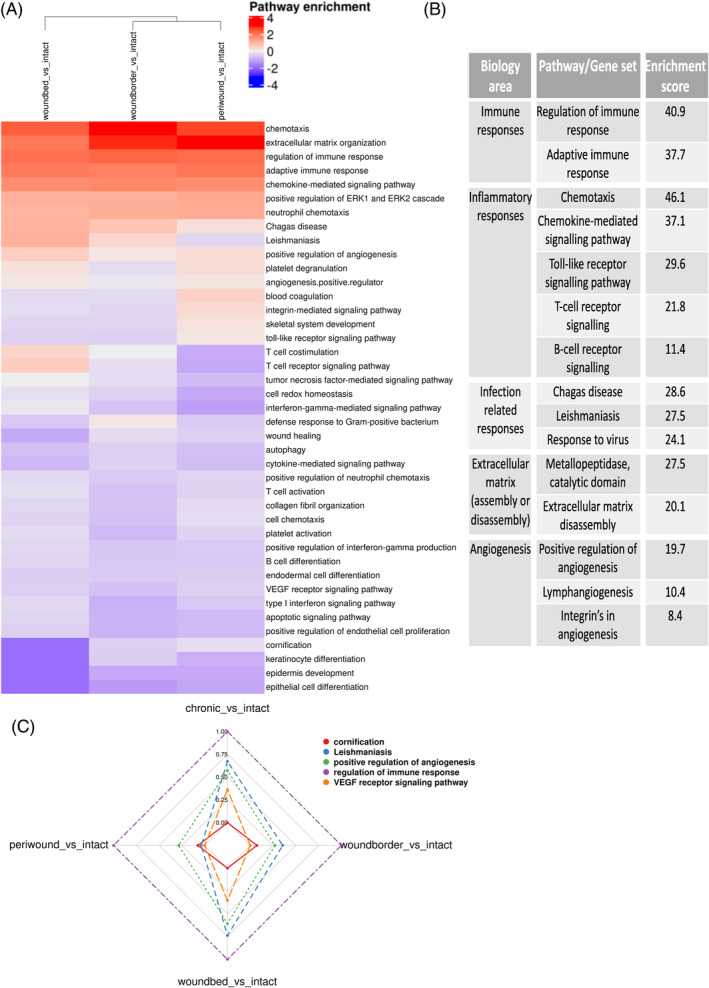
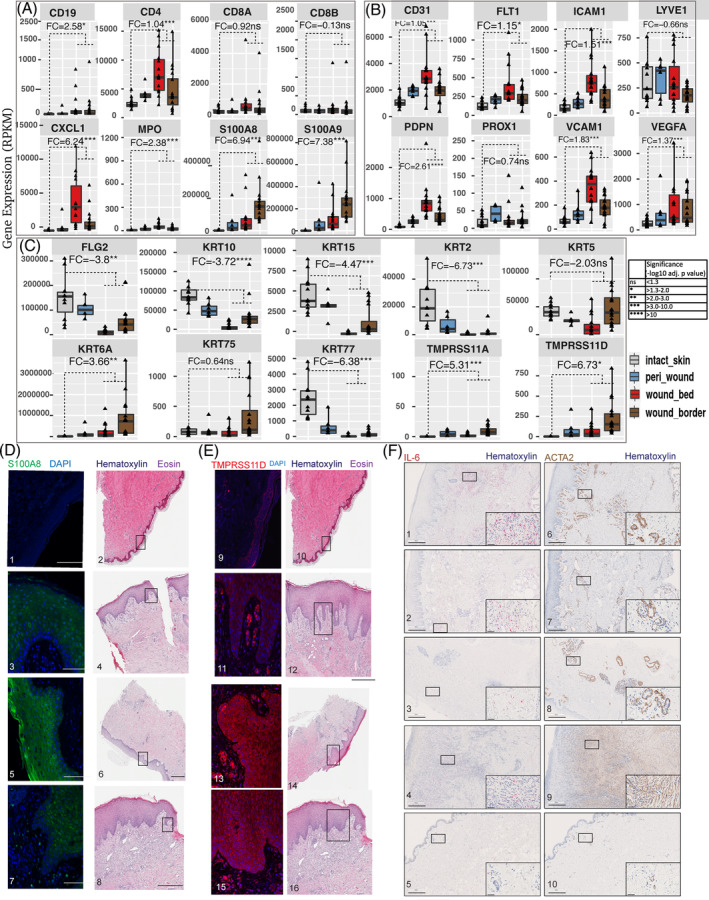
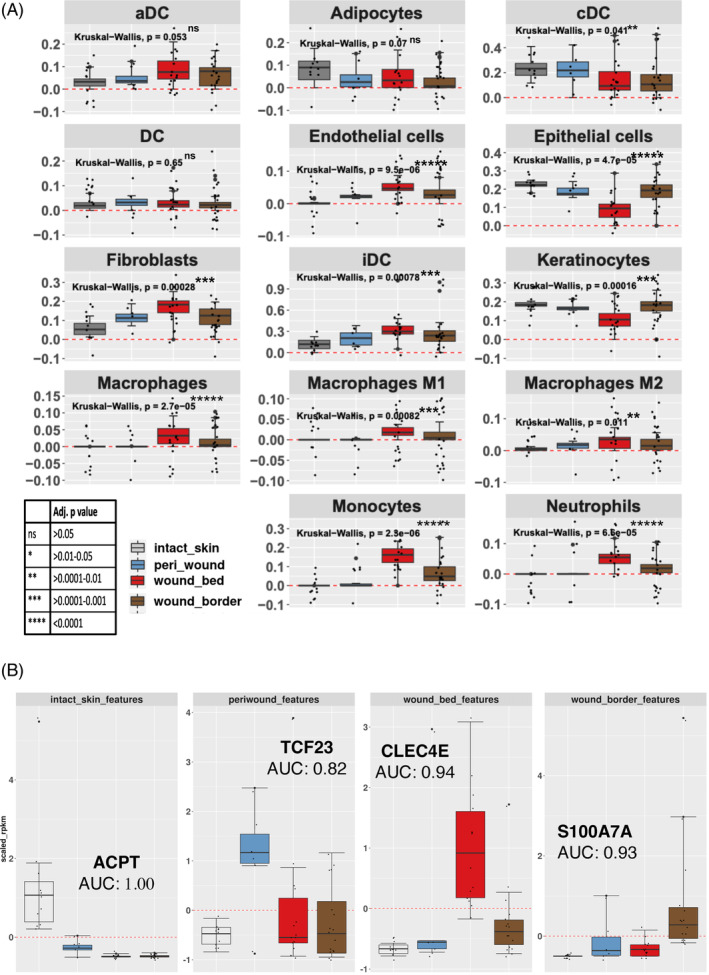
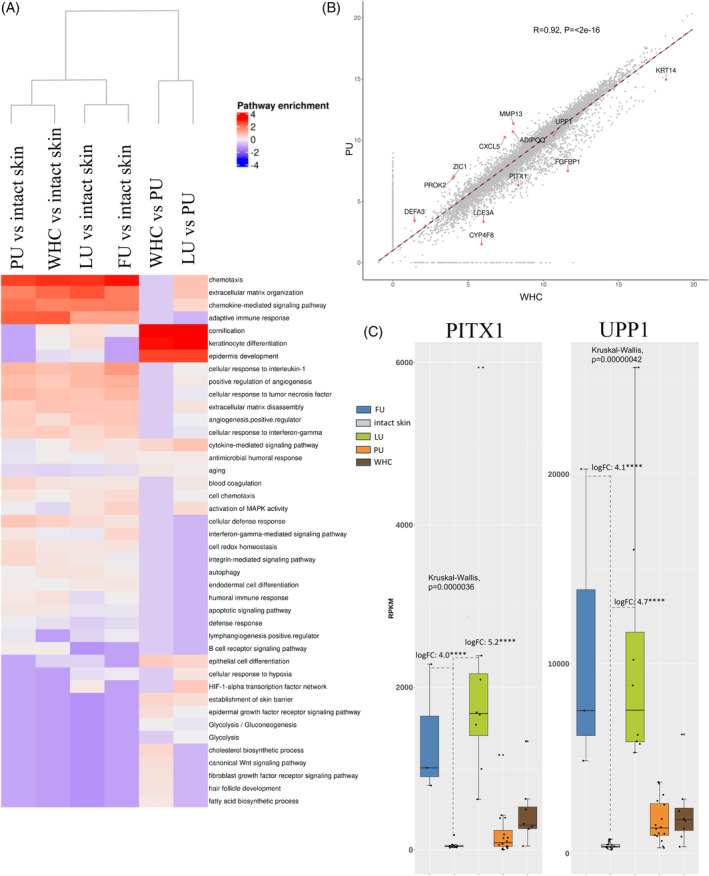
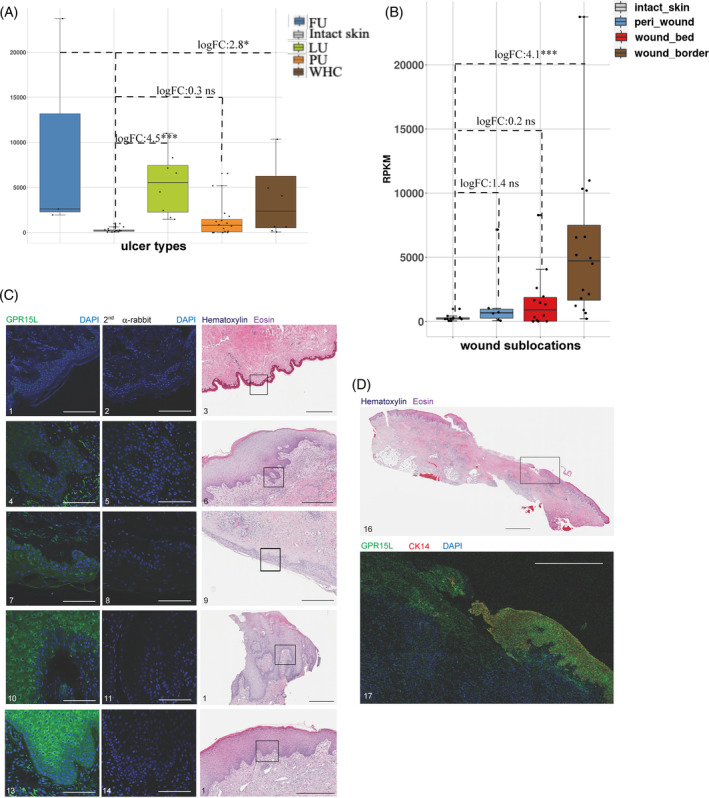
A limited understanding of the pathology underlying chronic wounds has hindered the development of effective diagnostic markers and pharmaceutical interventions. This study aimed to elucidate the molecular composition of various common chronic ulcer types to facilitate drug discovery strategies. We conducted a comprehensive analysis of leg ulcers (LUs), encompassing venous and arterial ulcers, foot ulcers (FUs), pressure ulcers (PUs), and compared them with surgical wound healing complications (WHCs). To explore the pathophysiological mechanisms and identify similarities or differences within wounds, we dissected wounds into distinct subregions, including the wound bed, border, and peri-wound areas, and compared them against intact skin. By correlating histopathology, RNA sequencing (RNA-Seq), and immunohistochemistry (IHC), we identified unique genes, pathways, and cell type abundance patterns in each wound type and subregion. These correlations aim to aid clinicians in selecting targeted treatment options and informing the design of future preclinical and clinical studies in wound healing. Notably, specific genes, such as PITX1 and UPP1, exhibited exclusive upregulation in LUs and FUs, potentially offering significant benefits to specialists in limb preservation and clinical treatment decisions. In contrast, comparisons between different wound subregions, regardless of wound type, revealed distinct expression profiles. The pleiotropic chemokine-like ligand GPR15L (C10orf99) and transmembrane serine proteases TMPRSS11A/D were significantly upregulated in wound border subregions. Interestingly, WHCs exhibited a nearly identical transcriptome to PUs, indicating clinical relevance. Histological examination revealed blood vessel occlusions with impaired angiogenesis in chronic wounds, alongside elevated expression of genes and immunoreactive markers related to blood vessel and lymphatic epithelial cells in wound bed subregions. Additionally, inflammatory and epithelial markers indicated heightened inflammatory responses in wound bed and border subregions and reduced wound bed epithelialization. In summary, chronic wounds from diverse anatomical sites share common aspects of wound pathophysiology but also exhibit distinct molecular differences. These unique molecular characteristics present promising opportunities for drug discovery and treatment, particularly for patients suffering from chronic wounds. The identified diagnostic markers hold the potential to enhance preclinical and clinical trials in the field of wound healing.
Keywords: RNA sequencing; chronic wounds; histopathology; quantitative immunohistochemistry; wound subregions.
© 2023 Novartis Biomedical Research and The Authors. International Wound Journal published by Medicalhelplines.com Inc and John Wiley & Sons Ltd.
Conflict of interest statement
All authors contributing to this manuscript declare that they have nothing to disclose and do not have a conflict of interest. All authors except D.W., M.V.S., D.S., B.W. and R.W., were or are employees of Novartis Pharma AG.
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
