

Acute Lymphoblastic Leukemia with Myeloid Mutations Is a High-Risk Disease Associated with Clonal Hematopoiesis
- PMID: 38150184
- PMCID: PMC11061587
- DOI: 10.1158/2643-3230.BCD-23-0106
Acute Lymphoblastic Leukemia with Myeloid Mutations Is a High-Risk Disease Associated with Clonal Hematopoiesis
Abstract
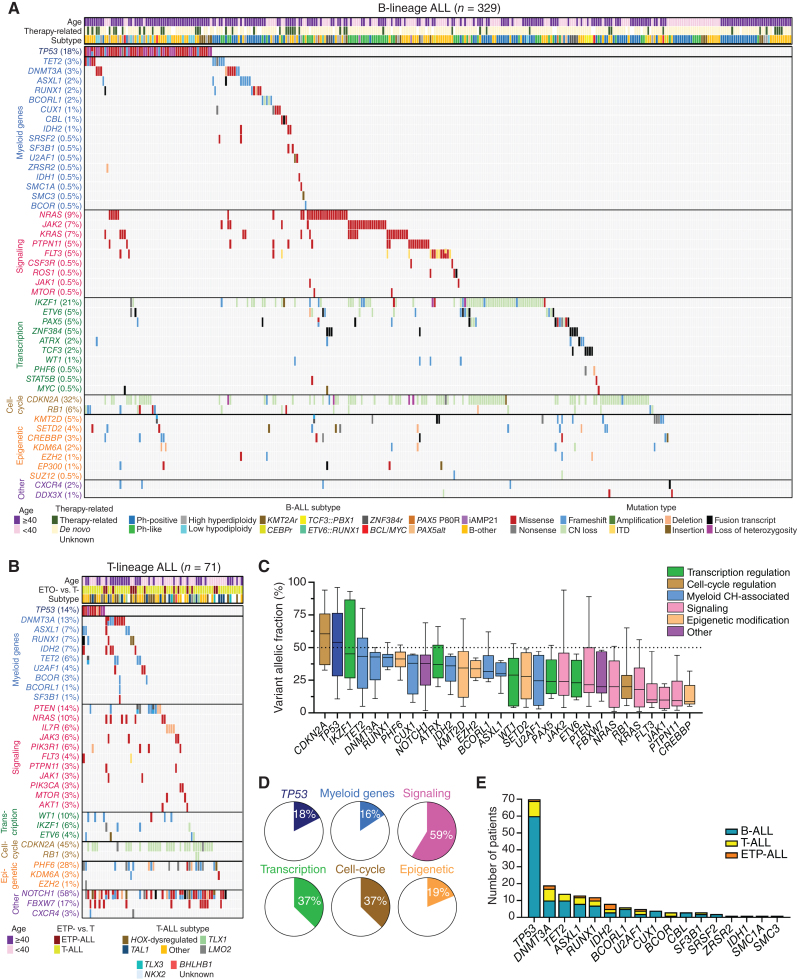
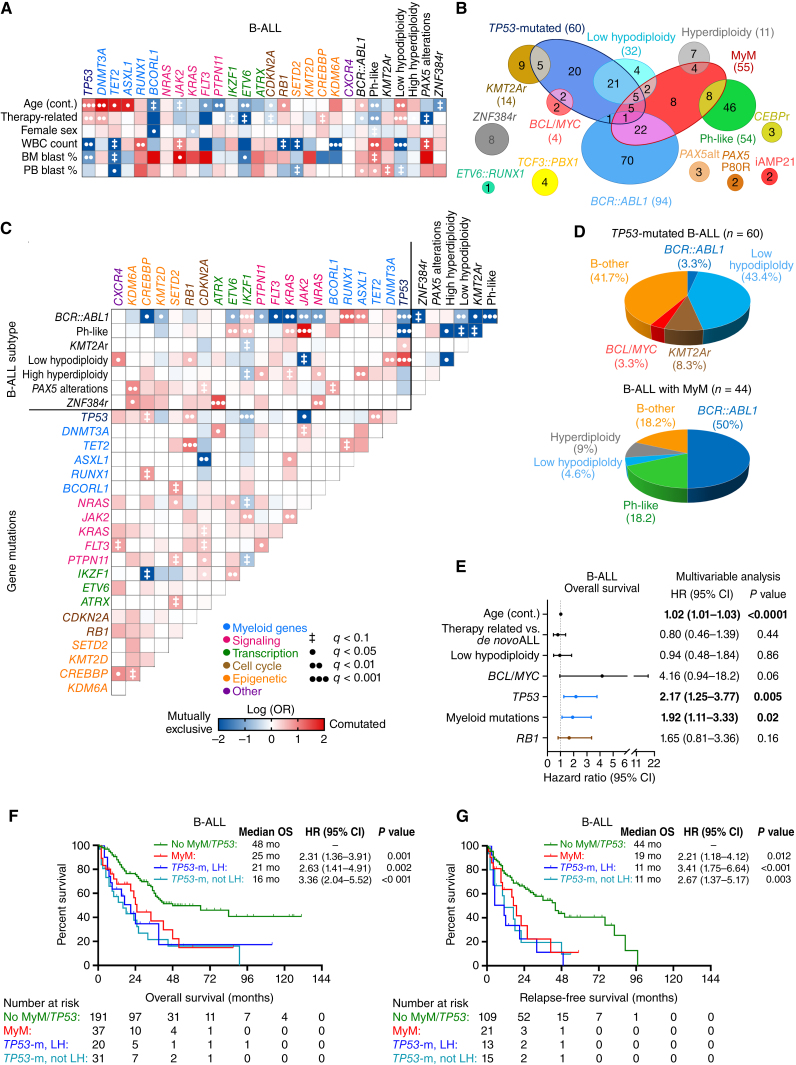
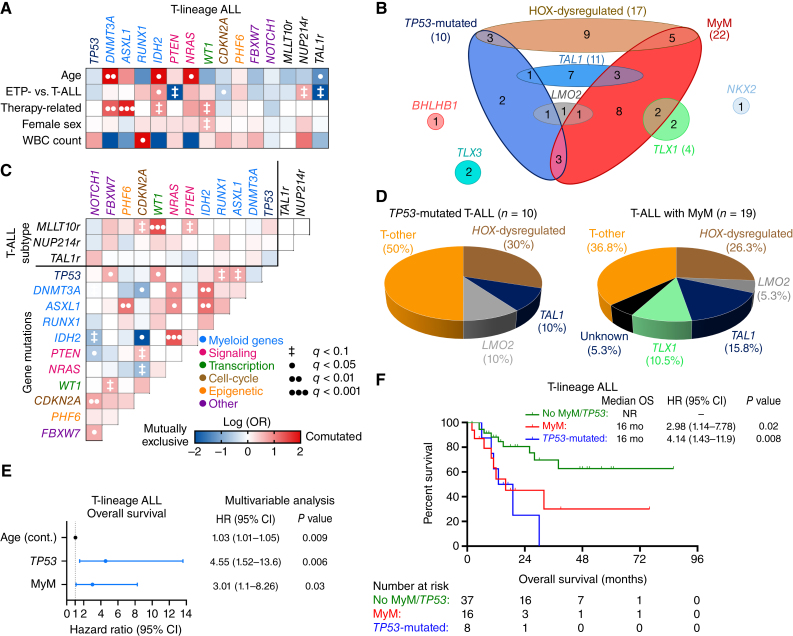
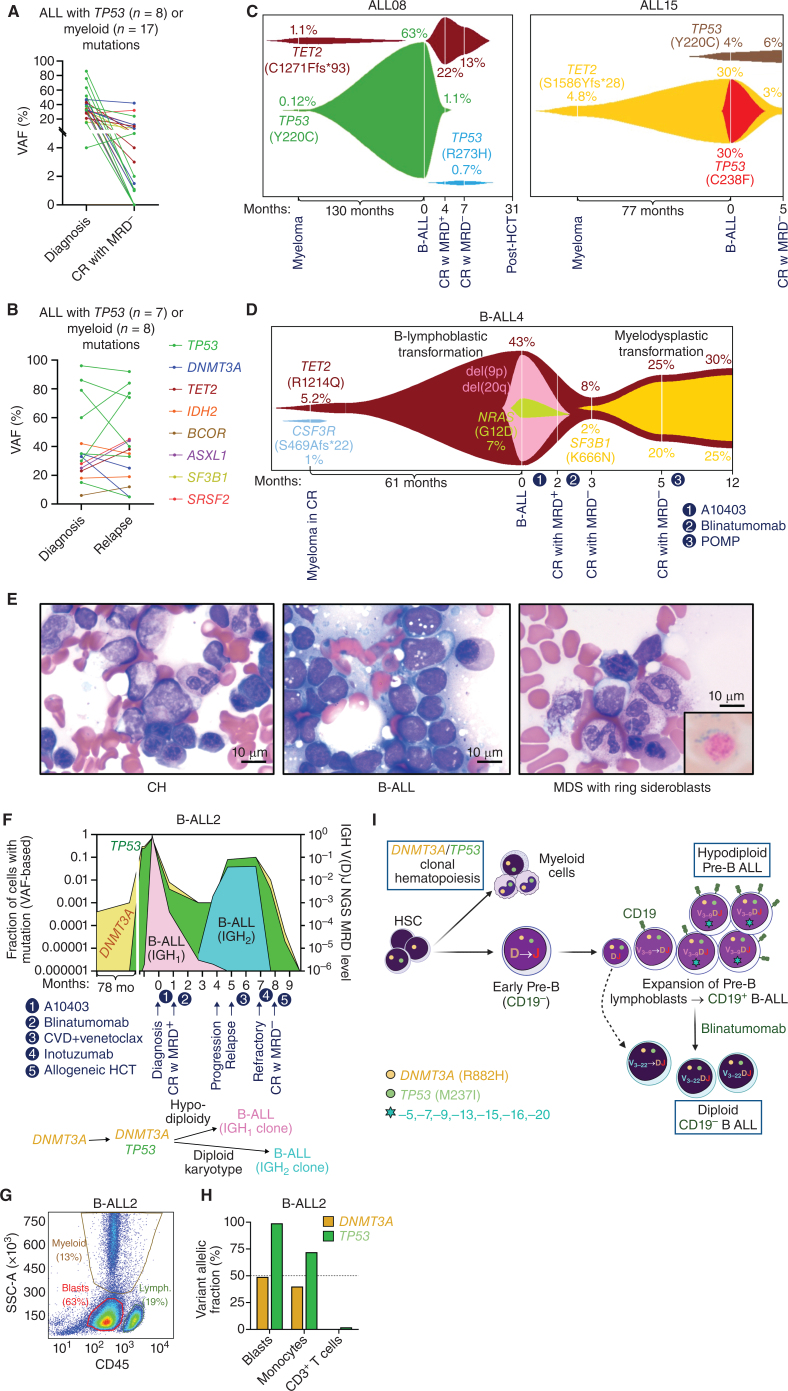
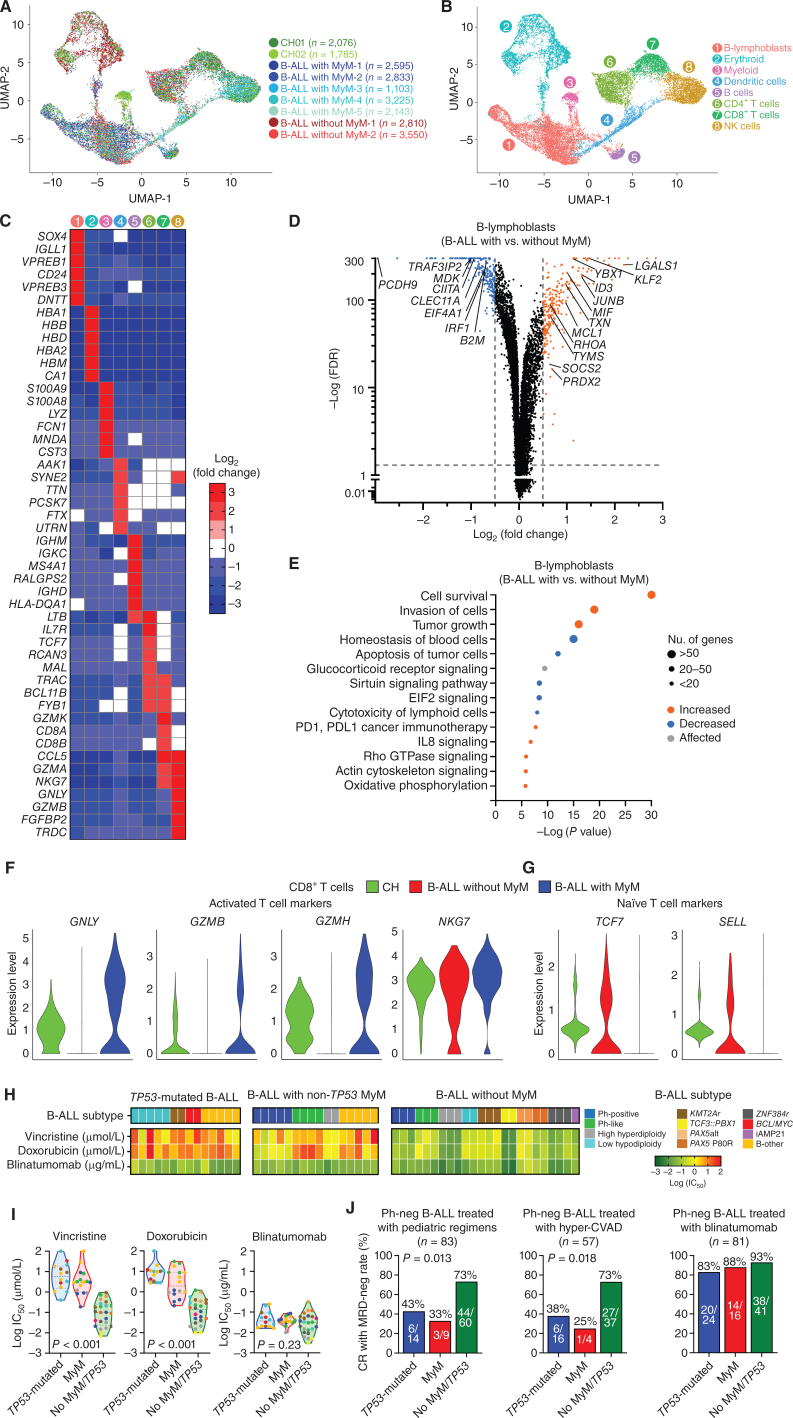
Myeloid neoplasms arise from preexisting clonal hematopoiesis (CH); however, the role of CH in the pathogenesis of acute lymphoblastic leukemia (ALL) is unknown. We found that 18% of adult ALL cases harbored TP53, and 16% had myeloid CH-associated gene mutations. ALL with myeloid mutations (MyM) had distinct genetic and clinical characteristics, associated with inferior survival. By using single-cell proteogenomic analysis, we demonstrated that myeloid mutations were present years before the diagnosis of ALL, and a subset of these clones expanded over time to manifest as dominant clones in ALL. Single-cell RNA sequencing revealed upregulation of genes associated with cell survival and resistance to apoptosis in B-ALL with MyM, which responds better to newer immunotherapeutic approaches. These findings define ALL with MyM as a high-risk disease that can arise from antecedent CH and offer new mechanistic insights to develop better therapeutic and preventative strategies.
Significance: CH is a precursor lesion for lymphoblastic leukemogenesis. ALL with MyM has distinct genetic and clinical characteristics, associated with adverse survival outcomes after chemotherapy. CH can precede ALL years before diagnosis, and ALL with MyM is enriched with activated T cells that respond to immunotherapies such as blinatumomab. See related commentary by Iacobucci, p. 142.
©2023 The Authors; Published by the American Association for Cancer Research.
Figures

References
-
- Brown PA, Shah B, Advani A, Aoun P, Boyer MW, Burke PW, et al. Acute lymphoblastic leukemia, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw 2021;19:1079–109. - PubMed
-
- Advani AS. Novel strategies in the treatment of acute lymphoblastic leukaemia. Lancet Haematol 2022;9:e240–e1. - PubMed
-
- Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin 2022;72:7–33. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
