

Clinical and genetic keys to cerebellar ataxia due to FGF14 GAA expansions
- PMID: 38150853
- PMCID: PMC10784672
- DOI: 10.1016/j.ebiom.2023.104931
Clinical and genetic keys to cerebellar ataxia due to FGF14 GAA expansions
Abstract
Background: SCA27B caused by FGF14 intronic heterozygous GAA expansions with at least 250 repeats accounts for 10-60% of cases with unresolved cerebellar ataxia. We aimed to assess the size and frequency of FGF14 expanded alleles in individuals with cerebellar ataxia as compared with controls and to characterize genetic and clinical variability.
Methods: We sized this repeat in 1876 individuals from France sampled for research purposes in this cross-sectional study: 845 index cases with cerebellar ataxia and 324 affected relatives, 475 controls, as well as 119 cases with spastic paraplegia, and 113 with familial essential tremor.
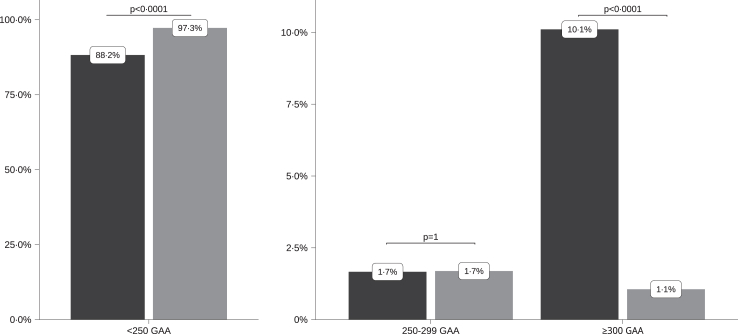
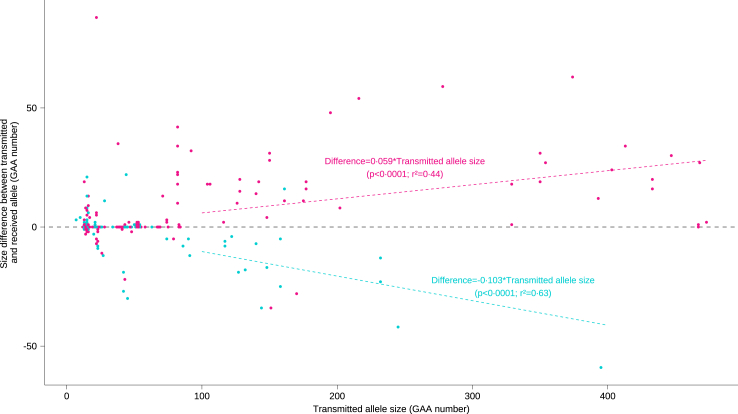
Findings: A higher frequency of expanded allele carriers in index cases with ataxia was significant only above 300 GAA repeats (10.1%, n = 85) compared with controls (1.1%, n = 5) (p < 0.0001) whereas GAA250-299 alleles were detected in 1.7% of both groups. Eight of 14 index cases with GAA250-299 repeats had other causal pathogenic variants (4/14) and/or discordance of co-segregation (5/14), arguing against GAA causality. We compared the clinical signs in 127 GAA≥300 carriers to cases with non-expanded GAA ataxia resulting in defining a key phenotype triad: onset after 45 years, downbeat nystagmus, episodic ataxic features including diplopia; and a frequent absence of dysarthria. All maternally transmitted alleles above 100 GAA were unstable with a median expansion of +18 repeats per generation (r2 = 0.44; p < 0.0001). In comparison, paternally transmitted alleles above 100 GAA mostly decreased in size (-15 GAA (r2 = 0.63; p < 0.0001)), resulting in the transmission bias observed in SCA27B pedigrees.
Interpretation: SCA27B diagnosis must consider both the phenotype and GAA expansion size. In carriers of GAA250-299 repeats, the absence of documented familial transmission and a presentation deviating from the key SCA27B phenotype, should prompt the search for an alternative cause. Affected fathers have a reduced risk of having affected children, which has potential implications for genetic counseling.
Funding: This work was supported by the Fondation pour la Recherche Médicale, grant number 13338 to JLM, the Association Connaître les Syndrome Cérébelleux - France (to GS) and by the European Union's Horizon 2020 research and innovation program under grant agreement No 779257 ("SOLVE-RD" to GS). DP holds a Fellowship award from the Canadian Institutes of Health Research (CIHR). SK received a grant (01GM1905C) from the Federal Ministry of Education and Research, Germany, through the TreatHSP network. This work was supported by the Australian Government National Health and Medical Research Council grants (GNT2001513 and MRFF2007677) to MB and PJL.
Keywords: Diagnosis; GAA expansion; Hereditary cerebellar ataxia; SCA27B.
Copyright © 2023 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests MA received consulting fees from Reata pharmaceuticals, Merz, AbbVie, Orkyn, Ever Pharma, Ipsen; honoraria from Reata pharmaceuticals, Merz, AbbVie, Orkyn, Ever Pharma, Ipsen and participated on an advisory board for Reata Pharmaceuticals. ILB received grants from Pfizer, Fondation Plan Alzheimer, JPND/ANR, Alector; consulting fees from Prevail Therapeutics, Alector, JEITO and participated on an advisory board of Prevail Therapeutics. CM reveived financial support for attending meetings and travel from Nutricia and participated on an advisory board of Medesis Pharma society. BD had public funding contracts for Clinical Research: Contrat de Recherche Clinique (CRC) 2021 (APHP), CRC 2023 (APHP) and Agence Régionale de Santé (ARS); received honoraria from MERZ, IPSEN Pharma, LVL Medical; received support for attending meetings from MERZ Pharma, ADELIA; participated on an advisory board of MERZ, ORION Pharma and received equipment from MERZ. SF received support for participation in national and international meetings from Alnylam. MB received honoraria for thesis examinations; is member of Australian Academy of Health and Medical Sciences Australian Learned Academies Data Internetworking Network (ALADIN) Project Steering Committee; Australian Academy of Health and Medical Sciences Reports Committee; Clinical Genomics Advisory Committee, Kinghorn Sequencing Center; Gen V Scientific Advisory Committee, Murdoch Children's Research Institute; Viertel Foundation Medical Advisory Board; Australian Academy of Health and Medical Sciences Reports Committee; Present American Epilepsy Society Basic Sciences Committee; Gen V Bioresource Genetics Working Group. AD received grants from Biogen, WAVELIFE, ROCHE, TRIPLET Therapeutics, NIH RO1 (National Institute of Health), National Hospital Clinical Research Program; consulting fees from Wavelife science, ROCHE, TRIPLET Therapeutics, Pfizer, ASKBIO, Genome Quebec, VICO therapeutics; participated on an advisory board for REATA; is the president of the Société Francophone de Neurogénétique.
Figures

References
-
- Coarelli G., Coutelier M., Durr A. Autosomal dominant cerebellar ataxias: new genes and progress towards treatments. Lancet Neurol. 2023;22:735–749. - PubMed
-
- Klockgether T., Mariotti C., Paulson H.L. Spinocerebellar ataxia. Nat Rev Dis Primer. 2019;5:24. - PubMed
-
- Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol. 2010;9:885–894. - PubMed
-
- Online mendelian inheritance in man. 2023. https://omim.org/
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
