

Metabolic Communication by SGLT2 Inhibition
- PMID: 38152989
- PMCID: PMC10922673
- DOI: 10.1161/CIRCULATIONAHA.123.065517
Metabolic Communication by SGLT2 Inhibition
Abstract
Background: SGLT2 (sodium-glucose cotransporter 2) inhibitors (SGLT2i) can protect the kidneys and heart, but the underlying mechanism remains poorly understood.
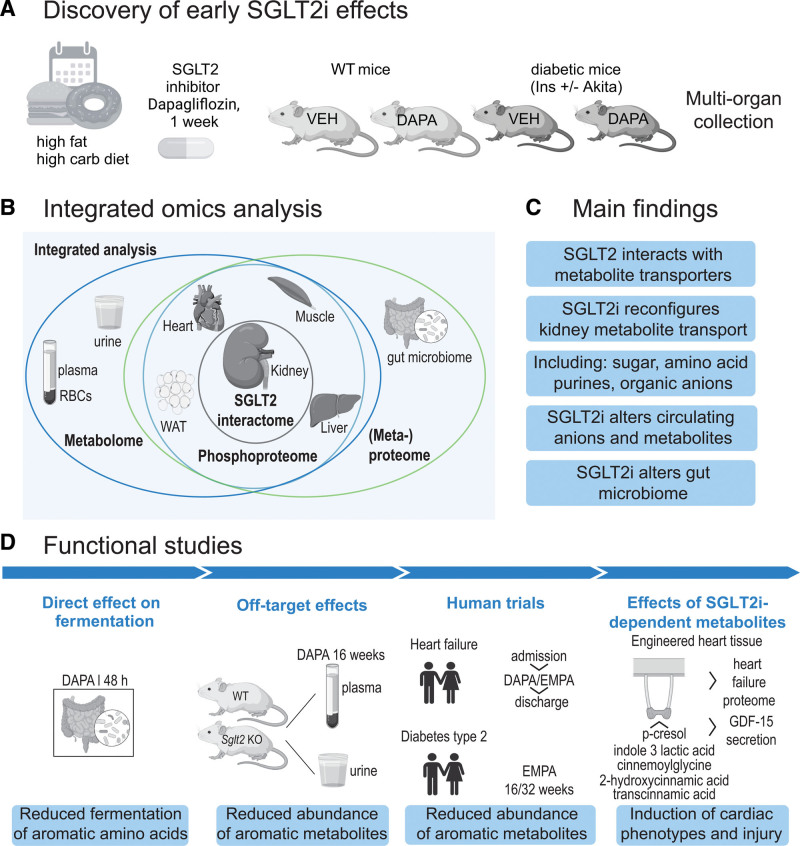
Methods: To gain insights on primary effects of SGLT2i that are not confounded by pathophysiologic processes or are secondary to improvement by SGLT2i, we performed an in-depth proteomics, phosphoproteomics, and metabolomics analysis by integrating signatures from multiple metabolic organs and body fluids after 1 week of SGLT2i treatment of nondiabetic as well as diabetic mice with early and uncomplicated hyperglycemia.
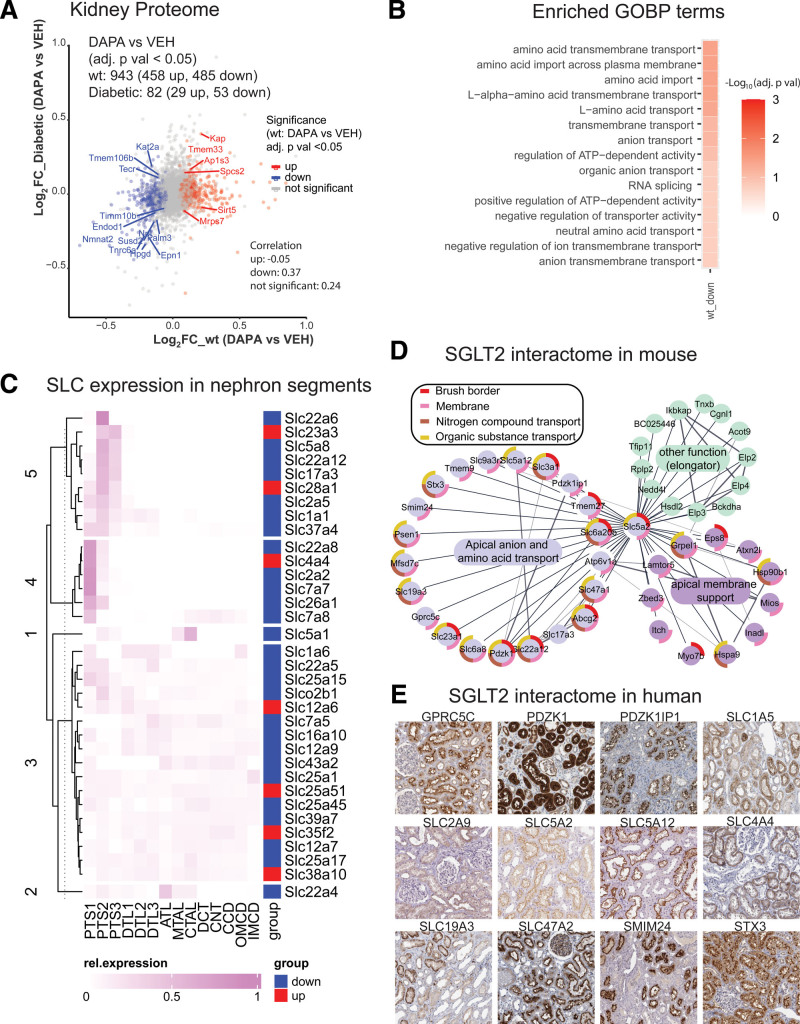
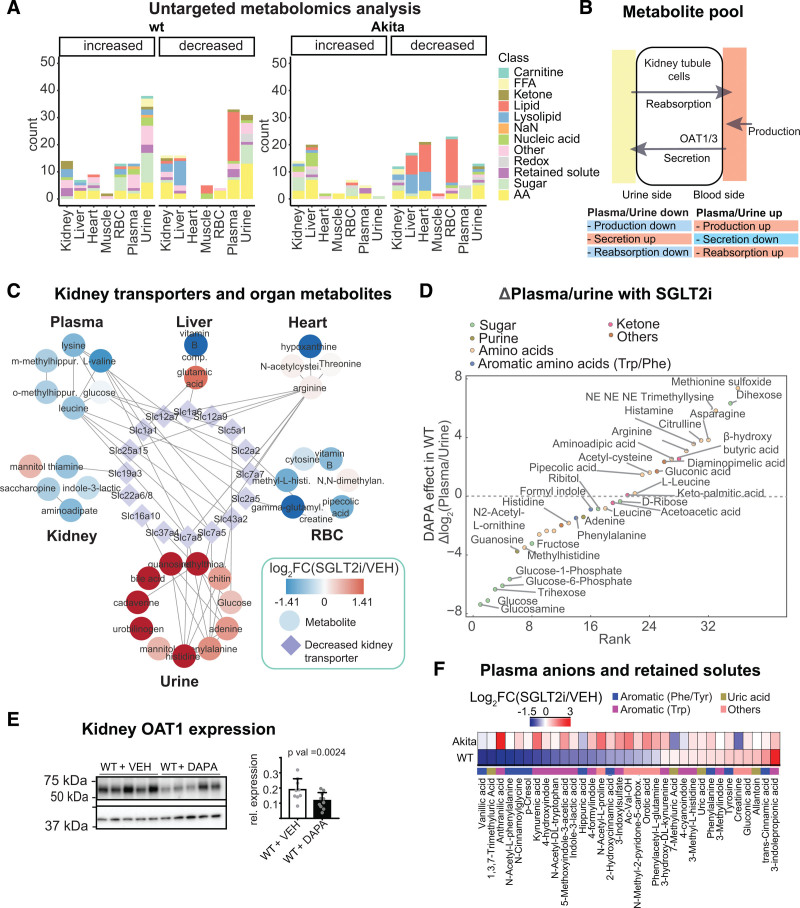
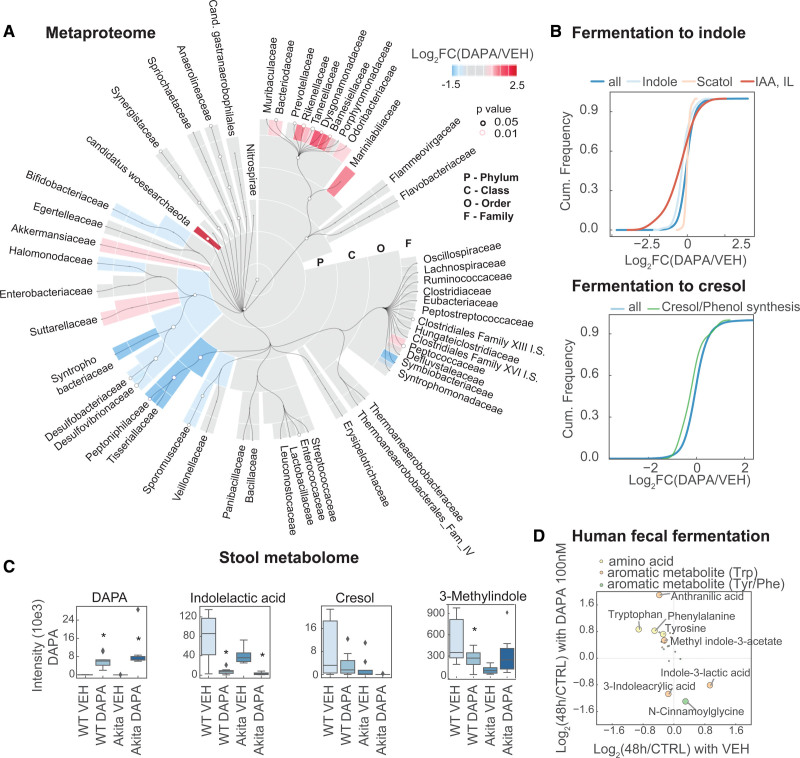
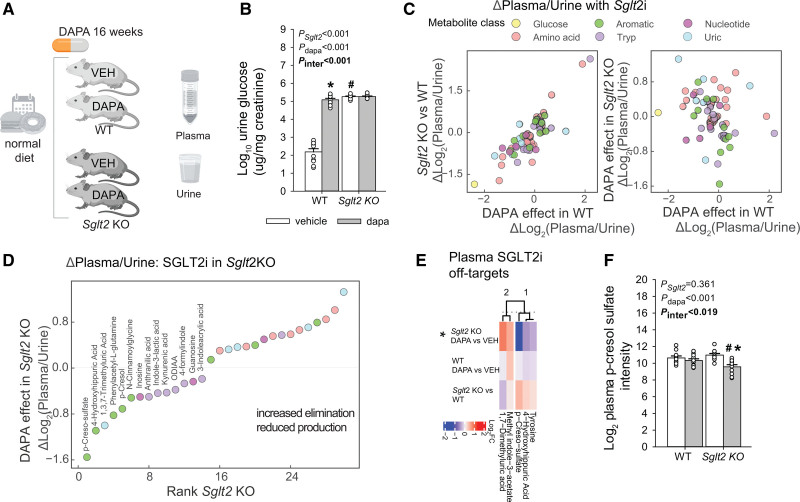
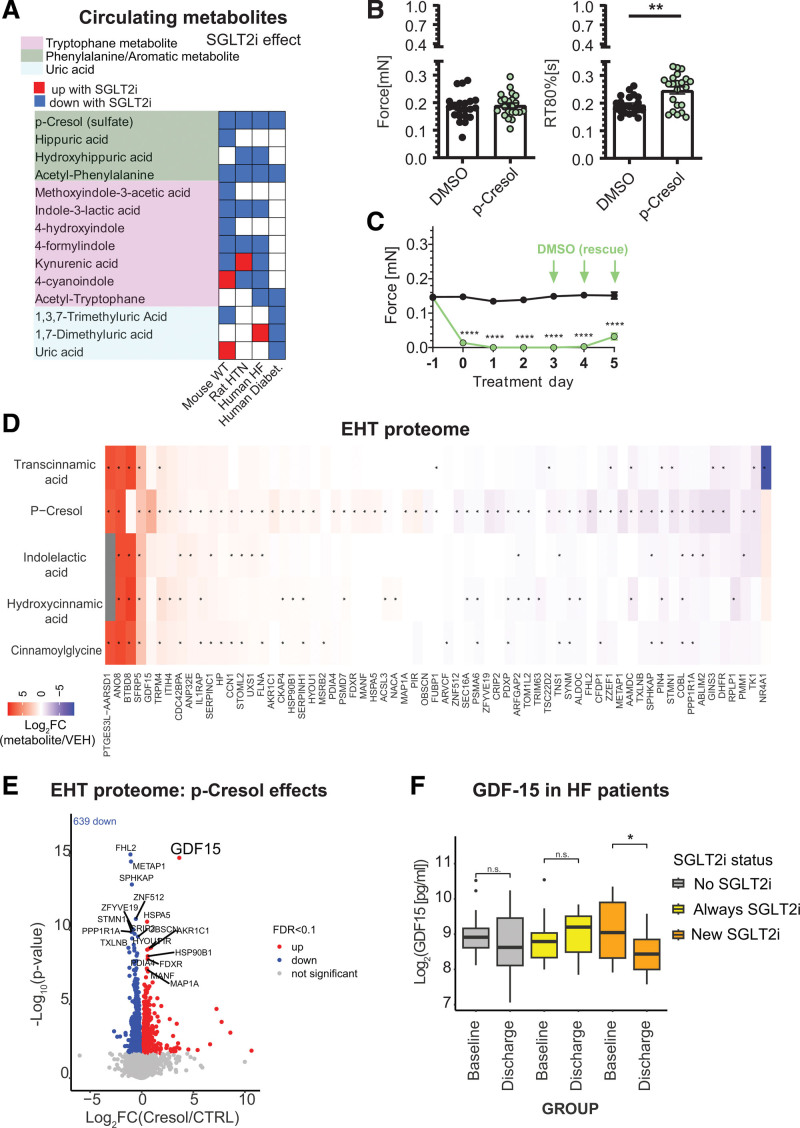
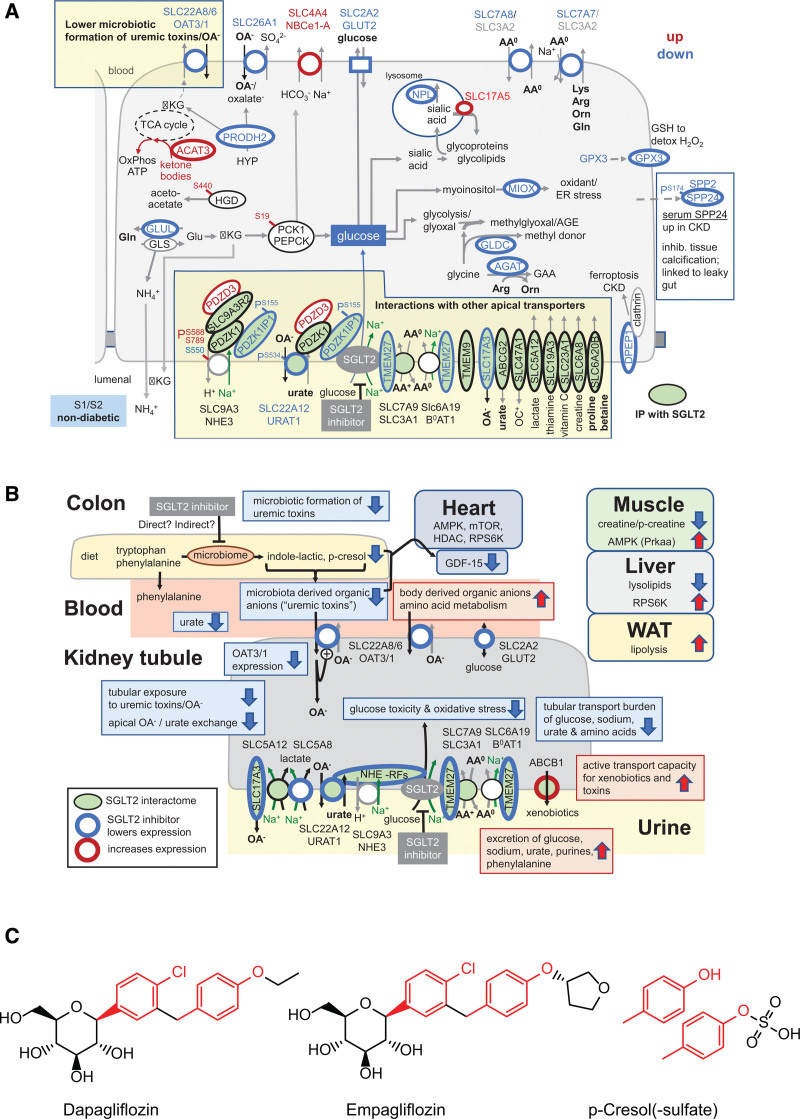
Results: Kidneys of nondiabetic mice reacted most strongly to SGLT2i in terms of proteomic reconfiguration, including evidence for less early proximal tubule glucotoxicity and a broad downregulation of the apical uptake transport machinery (including sodium, glucose, urate, purine bases, and amino acids), supported by mouse and human SGLT2 interactome studies. SGLT2i affected heart and liver signaling, but more reactive organs included the white adipose tissue, showing more lipolysis, and, particularly, the gut microbiome, with a lower relative abundance of bacteria taxa capable of fermenting phenylalanine and tryptophan to cardiovascular uremic toxins, resulting in lower plasma levels of these compounds (including p-cresol sulfate). SGLT2i was detectable in murine stool samples and its addition to human stool microbiota fermentation recapitulated some murine microbiome findings, suggesting direct inhibition of fermentation of aromatic amino acids and tryptophan. In mice lacking SGLT2 and in patients with decompensated heart failure or diabetes, the SGLT2i likewise reduced circulating p-cresol sulfate, and p-cresol impaired contractility and rhythm in human induced pluripotent stem cell-derived engineered heart tissue.
Conclusions: SGLT2i reduced microbiome formation of uremic toxins such as p-cresol sulfate and thereby their body exposure and need for renal detoxification, which, combined with direct kidney effects of SGLT2i, including less proximal tubule glucotoxicity and a broad downregulation of apical transporters (including sodium, amino acid, and urate uptake), provides a metabolic foundation for kidney and cardiovascular protection.
Keywords: diabetes mellitus; gastrointestinal microbiome; heart; kidney; metabolome; plasma; proteome; sodium-glucose transporter 2 inhibitors; uremic toxins; urine.
Conflict of interest statement
Figures

Comment in
-
Effects of SGLT2 inhibitors on the metabolic environment and uraemic toxins.Nat Rev Nephrol. 2024 Mar;20(3):155. doi: 10.1038/s41581-024-00814-4. Nat Rev Nephrol. 2024. PMID: 38291107 No abstract available.
-
Unexpected metabolic effects of sodium-glucose cotransporter 2 inhibitors.Kidney Int. 2024 Jul;106(1):12-15. doi: 10.1016/j.kint.2024.03.005. Epub 2024 Apr 10. Kidney Int. 2024. PMID: 38775770 No abstract available.
References
-
- Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Böhm M, Brunner-La Rocca H-P, Choi D-J, Chopra V, Chuquiure-Valenzuela E, et al. ; EMPEROR-Preserved Trial Investigators. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. 2021;385:1451–1461. doi: 10.1056/NEJMoa2107038 - PubMed
-
- Heerspink HJL, Stefánsson BV, Correa-Rotter R, Chertow GM, Greene T, Hou F-F, Mann JFE, McMurray JJV, Lindberg M, Rossing P, et al. ; DAPA-CKD Trial Committees and Investigators. Dapagliflozin in patients with chronic kidney disease. N Engl J Med. 2020;383:1436–1446. doi: 10.1056/NEJMoa2024816 - PubMed
-
- van der Aart–van der Beek AB, de Boer RA, Heerspink HJL. Kidney and heart failure outcomes associated with SGLT2 inhibitor use. Nat Rev Nephrol. 2022;18:294–306. doi: 10.1038/s41581-022-00535-6 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
