

Developmental trajectories and cooperating genomic events define molecular subtypes of BCR::ABL1-positive ALL
- PMID: 38153913
- PMCID: PMC11033585
- DOI: 10.1182/blood.2023021752
Developmental trajectories and cooperating genomic events define molecular subtypes of BCR::ABL1-positive ALL
Abstract
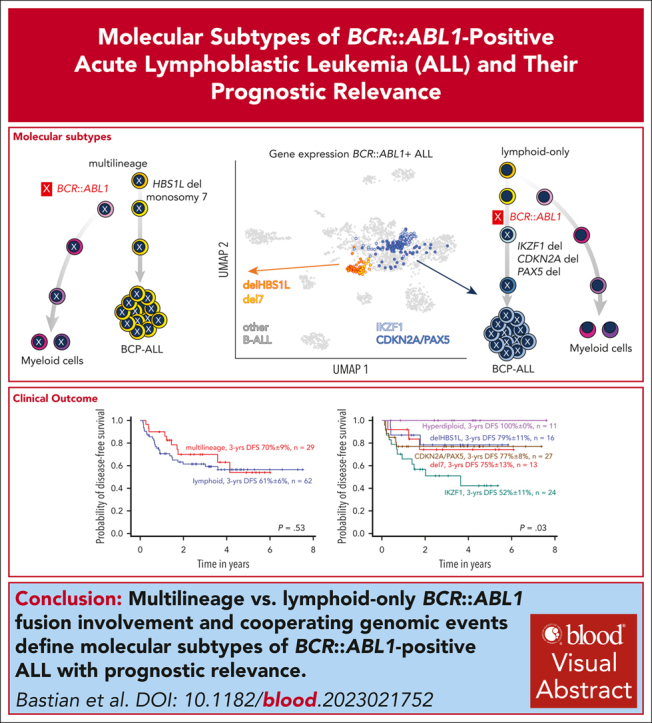
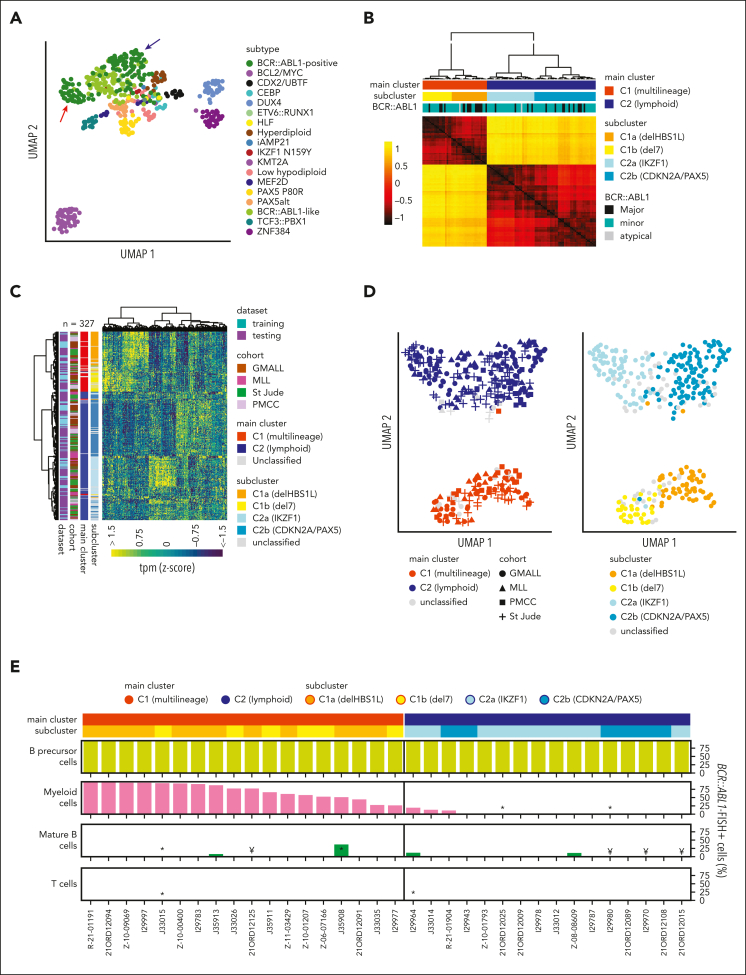
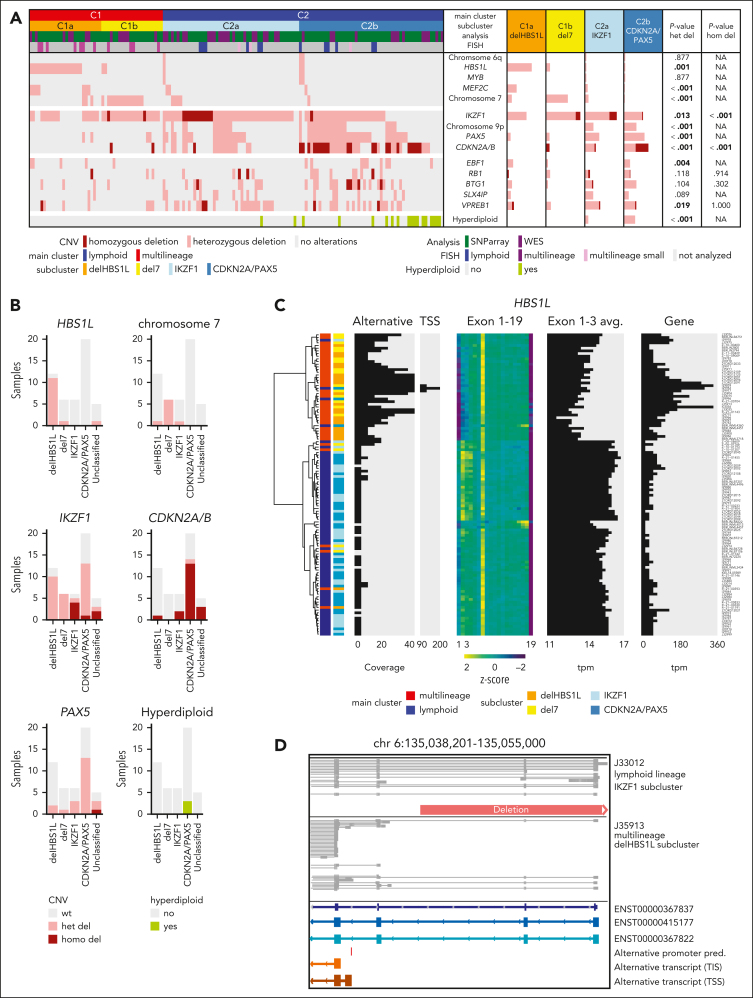
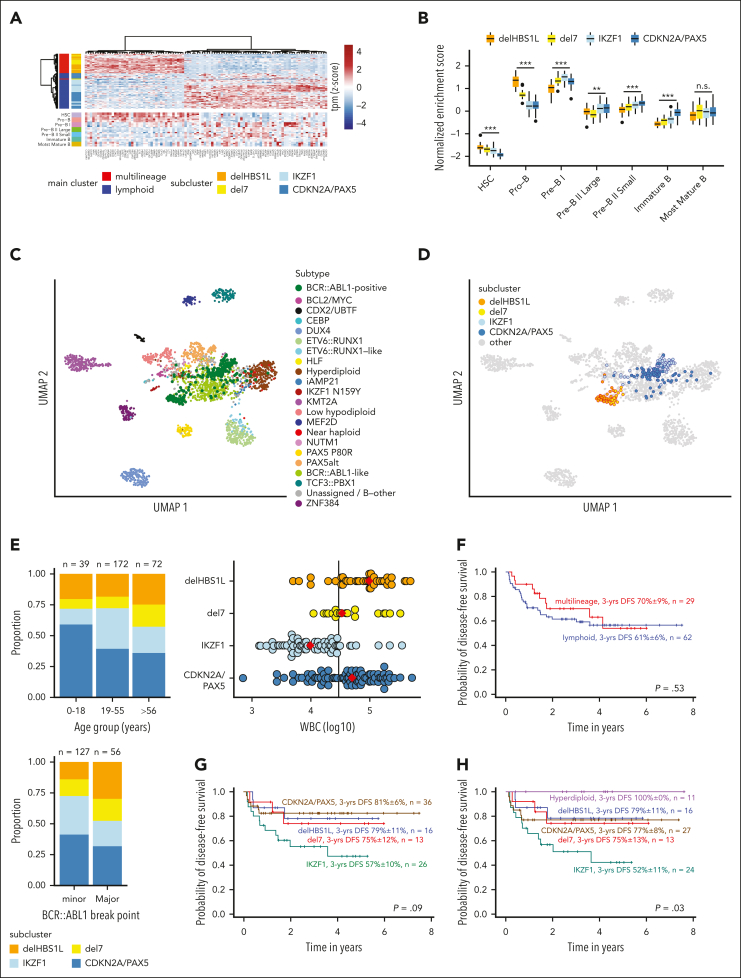
Distinct diagnostic entities within BCR::ABL1-positive acute lymphoblastic leukemia (ALL) are currently defined by the International Consensus Classification of myeloid neoplasms and acute leukemias (ICC): "lymphoid only", with BCR::ABL1 observed exclusively in lymphatic precursors, vs "multilineage", where BCR::ABL1 is also present in other hematopoietic lineages. Here, we analyzed transcriptomes of 327 BCR::ABL1-positive patients with ALL (age, 2-84 years; median, 46 years) and identified 2 main gene expression clusters reproducible across 4 independent patient cohorts. Fluorescence in situ hybridization analysis of fluorescence-activated cell-sorted hematopoietic compartments showed distinct BCR::ABL1 involvement in myeloid cells for these clusters (n = 18/18 vs n = 3/16 patients; P < .001), indicating that a multilineage or lymphoid BCR::ABL1 subtype can be inferred from gene expression. Further subclusters grouped samples according to cooperating genomic events (multilineage: HBS1L deletion or monosomy 7; lymphoid: IKZF1-/- or CDKN2A/PAX5 deletions/hyperdiploidy). A novel HSB1L transcript was highly specific for BCR::ABL1 multilineage cases independent of HBS1L genomic aberrations. Treatment on current German Multicenter Study Group for Adult ALL (GMALL) protocols resulted in comparable disease-free survival (DFS) for multilineage vs lymphoid cluster patients (3-year DFS: 70% vs 61%; P = .530; n = 91). However, the IKZF1-/- enriched lymphoid subcluster was associated with inferior DFS, whereas hyperdiploid cases showed a superior outcome. Thus, gene expression clusters define underlying developmental trajectories and distinct patterns of cooperating events in BCR::ABL1-positive ALL with prognostic relevance.
© 2024 American Society of Hematology. Published by Elsevier Inc. Licensed under Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International (CC BY-NC-ND 4.0), permitting only noncommercial, nonderivative use with attribution. All other rights reserved.
Conflict of interest statement
Conflict-of-interest disclosure: M.B. is contracted to perform research for Affimed, Amgen, and Regeneron; is a member of the advisory boards of Amgen and Incyte; and is on the speaker bureaus of Amgen, Janssen, Pfizer, and Roche. W.F. recieved personal fees and nonfinancial support from AbbVie; received grants, personal fees, and nonfinancial support from Amgen and Pfizer; received personal fees from Jazz Pharmaceuticals, Celgene, Morphosys, Ariad/Incyte, stem line therapeutics Daiichi Sankyo, Apis, Otsuka, and Servier outside the submitted work; has a patent issued for Amgen; and received support for medical writing from Amgen, Pfizer, and AbbVie. C.H. is part owner of the Munich Leukemia Laboratory. The remaining authors declare no competing financial interests.
Figures
Comment in
-
Uncovering new layers of Ph+ ALL biology.Blood. 2024 Apr 4;143(14):1322-1323. doi: 10.1182/blood.2023023583. Blood. 2024. PMID: 38573608 No abstract available.
References
-
- Pfeifer H, Wassmann B, Bethge W, et al. Randomized comparison of prophylactic and minimal residual disease-triggered imatinib after allogeneic stem cell transplantation for BCR–ABL1-positive acute lymphoblastic leukemia. Leukemia. 2013;27(6):1254–1262. - PubMed
-
- Pfeifer H, Lang F, Fiedler W, et al. P355: favorable outcome of Philadelphia-positive acute lymphoblastic leukemia with imatinib, dose-reduced induction followed by allogeneic stem cell transplantation –results from the GMALL trial 08/13. Hemasphere. 2023;7(S3)
-
- Martinelli G, Boissel N, Chevallier P, et al. Complete hematologic and molecular response in adult patients with relapsed/refractory Philadelphia chromosome–positive B-precursor acute lymphoblastic leukemia following treatment with blinatumomab: results from a phase II, single-arm, multicenter study. J Clin Oncol. 2017;35(16):1795–1802. - PubMed
-
- Foà R, Bassan R, Vitale A, et al. Dasatinib-blinatumomab for Ph-positive acute lymphoblastic leukemia in adults. N Engl J Med. 2020;383(17):1613–1623. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
