

Discovery of U2AF1 neoantigens in myeloid neoplasms
- PMID: 38164756
- PMCID: PMC10729103
- DOI: 10.1136/jitc-2023-007490
Discovery of U2AF1 neoantigens in myeloid neoplasms
Abstract
Background: Myelodysplastic syndromes (MDS) arise from somatic mutations acquired in hematopoietic stem and progenitor cells, causing cytopenias and predisposing to transformation into secondary acute myeloid leukemia (sAML). Recurrent mutations in spliceosome genes, including U2AF1, are attractive therapeutic targets as they are prevalent in MDS and sAML, arise early in neoplastic cells, and are generally absent from normal cells, including normal hematopoietic cells. MDS and sAML are susceptible to T cell-mediated killing, and thus engineered T-cell immunotherapies hold promise for their treatment. We hypothesized that targeting spliceosome mutation-derived neoantigens with transgenic T-cell receptor (TCR) T cells would selectively eradicate malignant cells in MDS and sAML.
Methods: We identified candidate neoantigen epitopes from recurrent protein-coding mutations in the spliceosome genes SRSF2 and U2AF1 using a multistep in silico process. Candidate epitopes predicted to bind human leukocyte antigen (HLA) class I, be processed and presented from the parent protein, and not to be subject to tolerance then underwent in vitro immunogenicity screening. CD8+ T cells recognizing immunogenic neoantigen epitopes were evaluated in in vitro assays to assess functional avidity, confirm the predicted HLA restriction, the potential for recognition of similar peptides, and the ability to kill neoplastic cells in an antigen-specific manner. Neoantigen-specific TCR were sequenced, cloned into lentiviral vectors, and transduced into third-party T cells after knock-out of endogenous TCR, then tested in vitro for specificity and ability to kill neoplastic myeloid cells presenting the neoantigen. The efficacy of neoantigen-specific T cells was evaluated in vivo in a murine cell line-derived xenograft model.
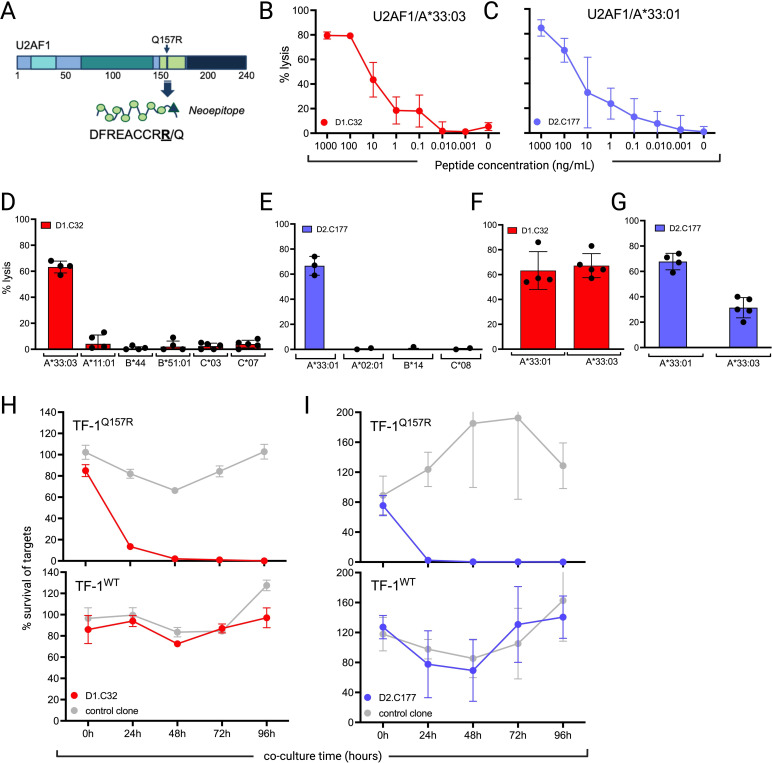
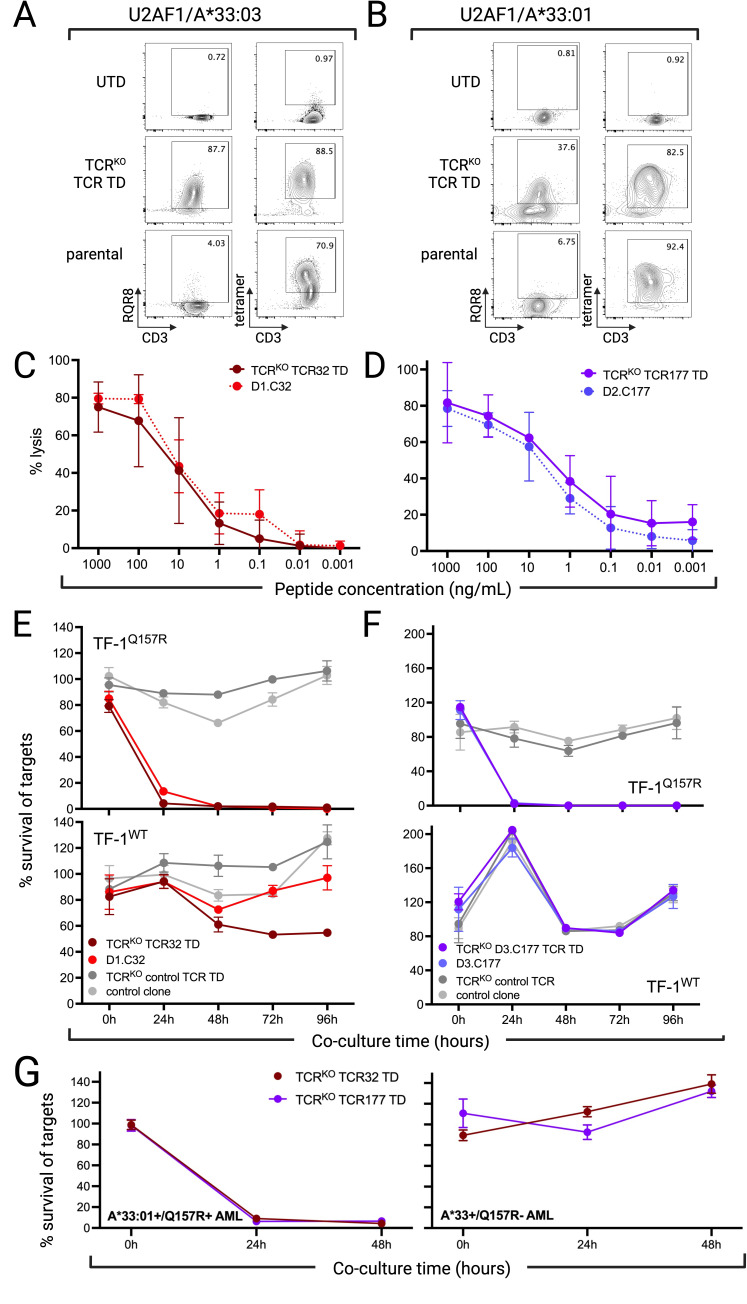
Results: We identified two neoantigens created from a recurrent mutation in U2AF1, isolated CD8+ T cells specific for the neoantigens, and demonstrated that transferring their TCR to third-party CD8+ T cells is feasible and confers specificity for the U2AF1 neoantigens. Finally, we showed that these neoantigen-specific TCR-T cells do not recognize normal hematopoietic cells but efficiently kill malignant myeloid cells bearing the specific U2AF1 mutation, including primary cells, in vitro and in vivo.
Conclusions: These data serve as proof-of-concept for developing precision medicine approaches that use neoantigen-directed T-cell receptor-transduced T cells to treat MDS and sAML.
Keywords: Antigens, Neoplasm; Hematologic Neoplasms; Immunotherapy, Adoptive.
© Author(s) (or their employer(s)) 2023. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: MB is a Founder and Scientific Advisory Board member of HighPassBio, and a Scientific Advisory Board member of Orca Bio, and has also received compensation from Miltenyi Biotec for presentations at conferences and corporate symposia pertaining to research unrelated to the current manuscript. MB and MAB have filed a provisional patent application number 63/274,681 covering applications of T cell immunotherapy for U2AF1-mutated malignancies.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous