

Clinical, Imaging, Genetic, and Disease Course Characteristics in Patients With GM2 Gangliosidosis: Beyond Age of Onset
- PMID: 38165373
- PMCID: PMC10834127
- DOI: 10.1212/WNL.0000000000207898
Clinical, Imaging, Genetic, and Disease Course Characteristics in Patients With GM2 Gangliosidosis: Beyond Age of Onset
Erratum in
-
Clinical, Imaging, Genetic, and Disease Course Characteristics in Patients With GM2 Gangliosidosis: Beyond Age of Onset.Neurology. 2024 May 14;102(9):e209406. doi: 10.1212/WNL.0000000000209406. Epub 2024 Apr 26. Neurology. 2024. PMID: 38669633 Free PMC article. No abstract available.
Abstract
Background and objectives: GM2 gangliosidoses, a group of autosomal-recessive neurodegenerative lysosomal storage disorders, result from β-hexosaminidase (HEX) deficiency with GM2 ganglioside as its main substrate. Historically, GM2 gangliosidoses have been classified into infantile, juvenile, and late-onset forms. With disease-modifying treatment trials now on the horizon, a more fine-grained understanding of the disease course is needed.
Methods: We aimed to map and stratify the clinical course of GM2 gangliosidoses in a multicenter cohort of pediatric and adult patients. Patients were stratified according to age at onset and age at diagnosis. The 2 resulting GM2 disease clusters were characterized in-depth for respective disease features (detailed standardized clinical, laboratory, and MRI assessments) and disease evolution.
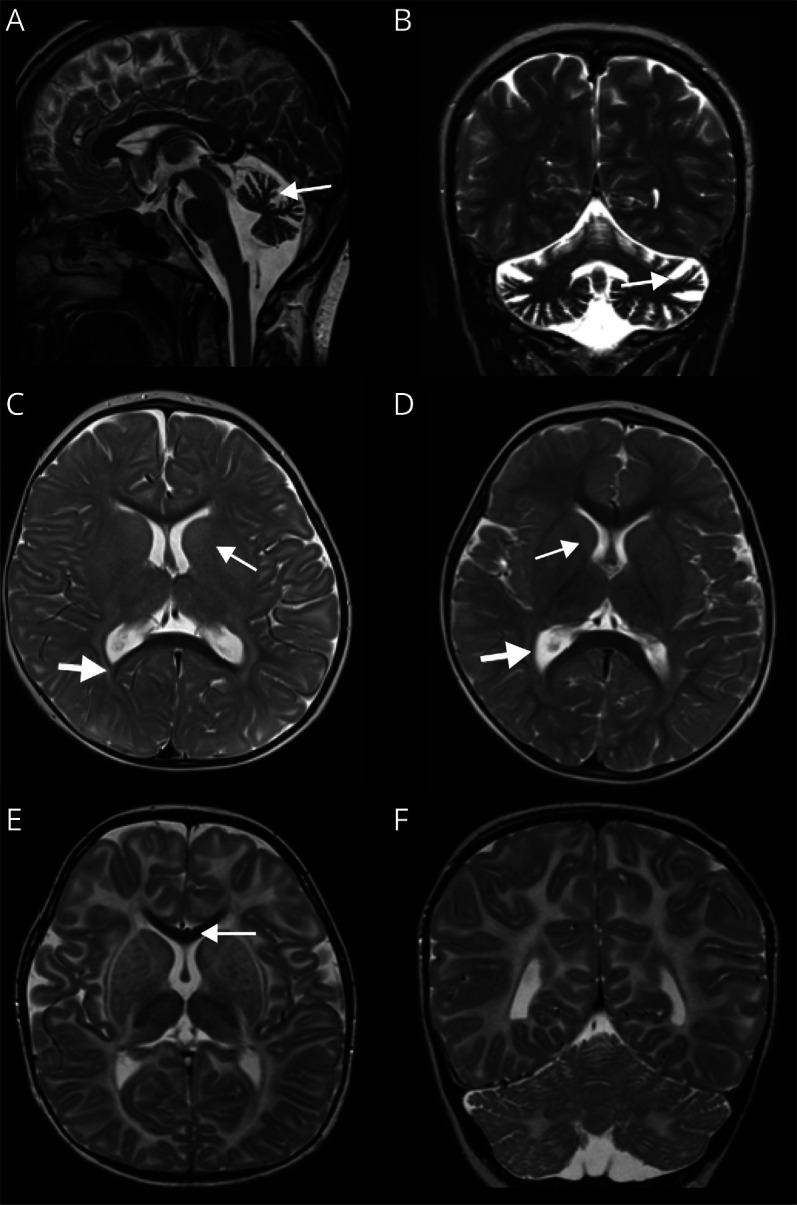
Results: In 21 patients with GM2 gangliosidosis (17 Tay-Sachs, 2 GM2 activator deficiency, 2 Sandhoff disease), 2 disease clusters were discriminated: an early-onset and early diagnosis cluster (type I; n = 8, including activator deficiency and Sandhoff disease) and a cluster with very variable onset and long interval until diagnosis (type II; n = 13 patients). In type I, rapid onset of developmental stagnation and regression, spasticity, and seizures dominated the clinical picture. Cherry red spot, startle reactions, and elevated AST were only seen in this cluster. In type II, problems with balance or gait, muscle weakness, dysarthria, and psychiatric symptoms were specific and frequent symptoms. Ocular signs were common, including supranuclear vertical gaze palsy in 30%. MRI involvement of basal ganglia and peritrigonal hyperintensity was seen only in type I, whereas predominant infratentorial atrophy (or normal MRI) was characteristic in type II. These types were, at least in part, associated with certain genetic variants.
Discussion: Age at onset alone seems not sufficient to adequately predict different disease courses in GM2 gangliosidosis, as required for upcoming trial planning. We propose an alternative classification based on age at disease onset and dynamics, predicted by clinical features and biomarkers, into type I-an early-onset, rapid progression cluster-and type II-a variable onset, slow progression cluster. Specific diagnostic workup, including GM2 gangliosidosis, should be performed in patients with combined ataxia plus lower motor neuron weakness to identify type II patients.
Conflict of interest statement
M. Synofzik has received consultancy honoraria from Ionis, UCB, Prevail, Orphazyme, Servier, Reata, GenOrph, AviadoBio, Biohaven, Zevra and Lilly, all unrelated to the present manuscript. J. Kern, J. Böhringer, D. Timmann, R. Trollmann, C. Stendel, C. Kamm, M. Röbl, V. Santhanakumaran, S. Groeschel, S. Beck-Wödl, S. Göricke, I. Krägeloh-Mann reports no disclosures relevant to the manuscript. Go to
Figures




References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources