

Cell cycle arrest induces lipid droplet formation and confers ferroptosis resistance
- PMID: 38167301
- PMCID: PMC10761718
- DOI: 10.1038/s41467-023-44412-7
Cell cycle arrest induces lipid droplet formation and confers ferroptosis resistance
Abstract
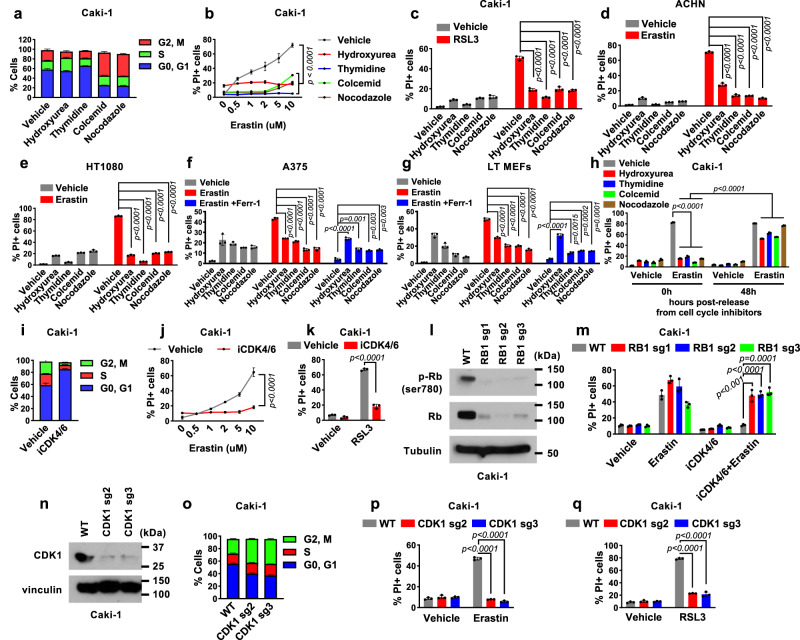
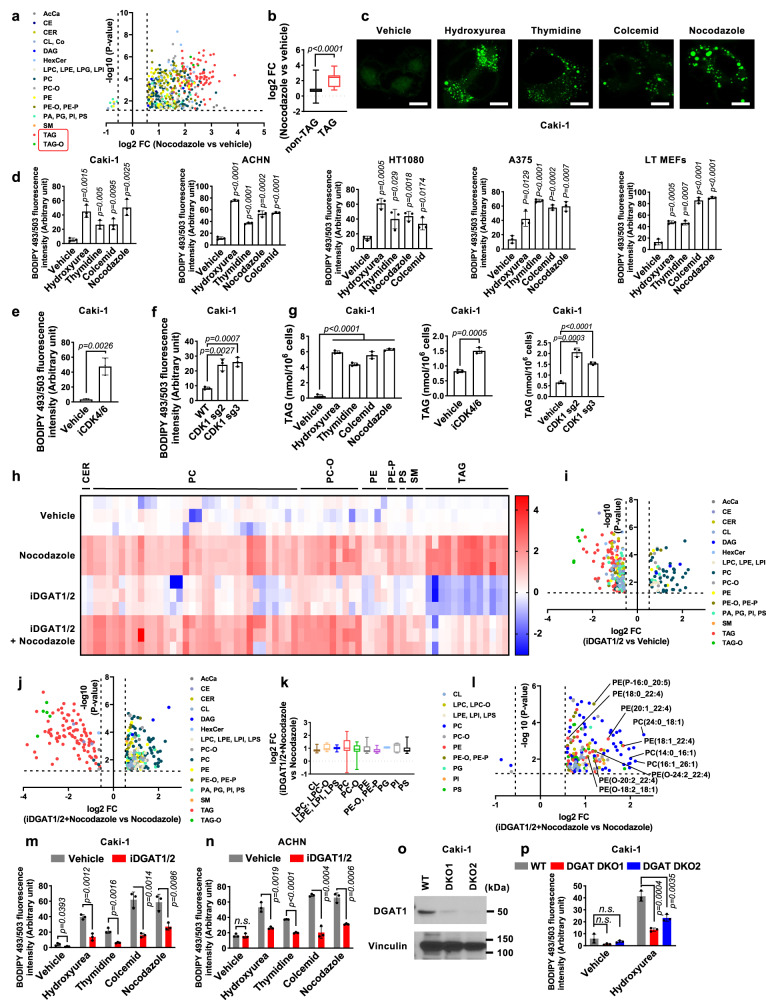
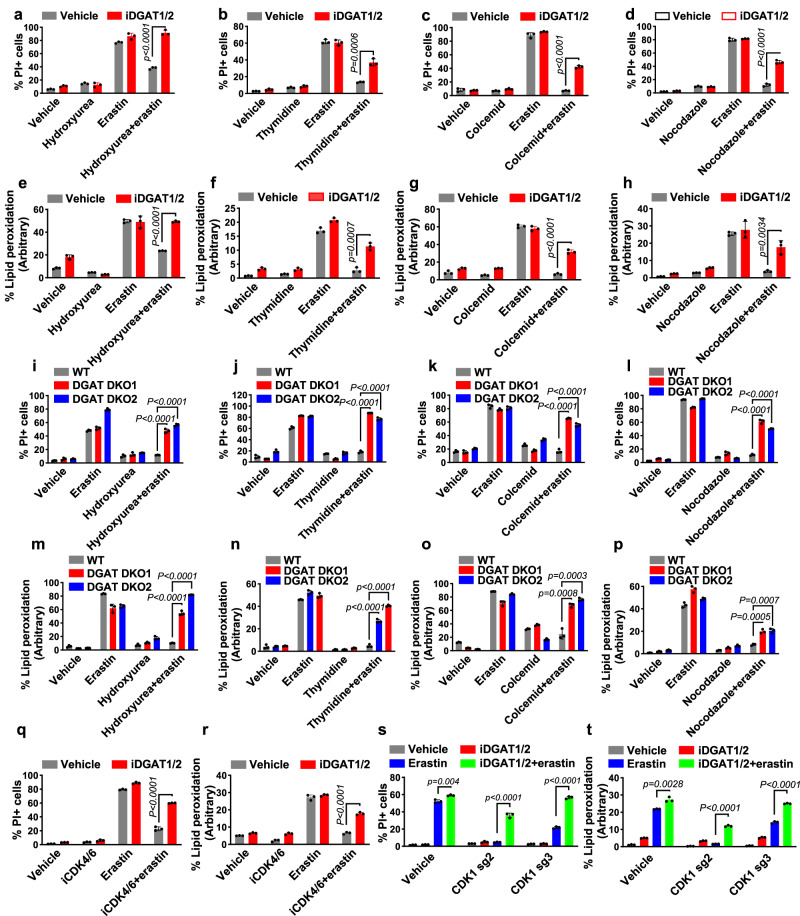
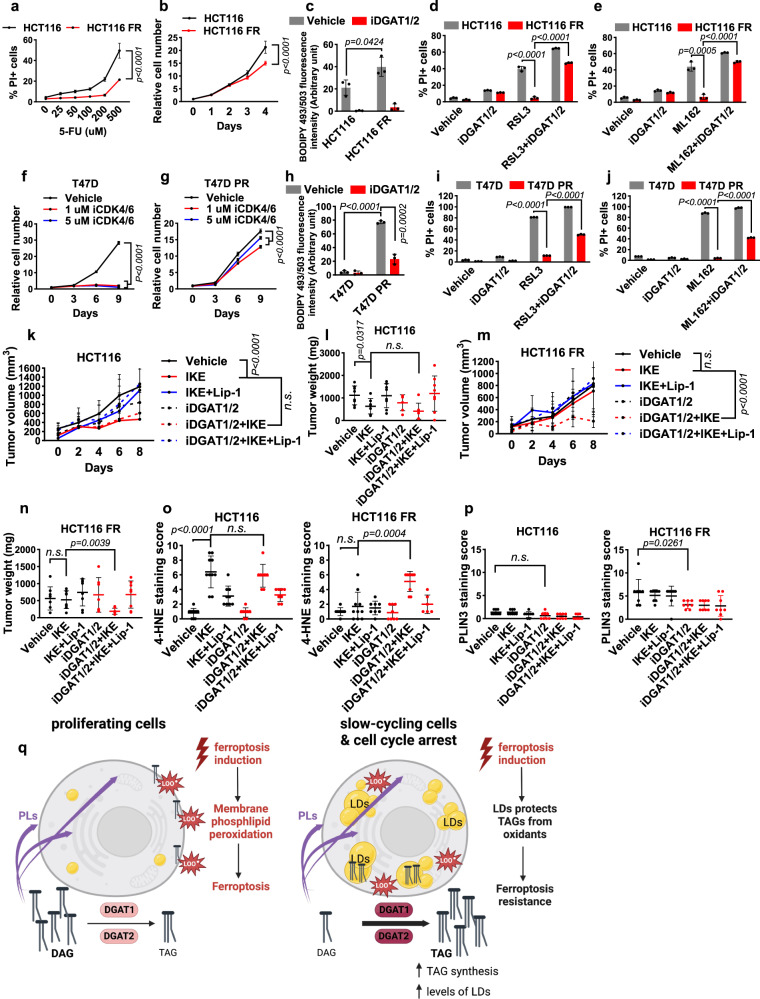
How cells coordinate cell cycling with cell survival and death remains incompletely understood. Here, we show that cell cycle arrest has a potent suppressive effect on ferroptosis, a form of regulated cell death induced by overwhelming lipid peroxidation at cellular membranes. Mechanistically, cell cycle arrest induces diacylglycerol acyltransferase (DGAT)-dependent lipid droplet formation to sequester excessive polyunsaturated fatty acids (PUFAs) that accumulate in arrested cells in triacylglycerols (TAGs), resulting in ferroptosis suppression. Consequently, DGAT inhibition orchestrates a reshuffling of PUFAs from TAGs to phospholipids and re-sensitizes arrested cells to ferroptosis. We show that some slow-cycling antimitotic drug-resistant cancer cells, such as 5-fluorouracil-resistant cells, have accumulation of lipid droplets and that combined treatment with ferroptosis inducers and DGAT inhibitors effectively suppresses the growth of 5-fluorouracil-resistant tumors by inducing ferroptosis. Together, these results reveal a role for cell cycle arrest in driving ferroptosis resistance and suggest a ferroptosis-inducing therapeutic strategy to target slow-cycling therapy-resistant cancers.
© 2024. The Author(s).
Conflict of interest statement
B.G. reports receiving consultation fees from Guidepoint Global, Cambridge Solutions, and NGM Bio, and is an inventor with patent applications involving targeting ferroptosis in cancer therapy. K.O. and L.K. are former full-time employees of Kadmon Corporation and are now full-time employees of the Carl Icahn Labs and Sanofi, US, respectively. M.V.P. is a former full-time employee of Kadmon Corporation and is now a full-time employee at PMV Pharmaceutics. The other authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
