

Histone lactylation couples cellular metabolism with developmental gene regulatory networks
- PMID: 38167340
- PMCID: PMC10762033
- DOI: 10.1038/s41467-023-44121-1
Histone lactylation couples cellular metabolism with developmental gene regulatory networks
Abstract
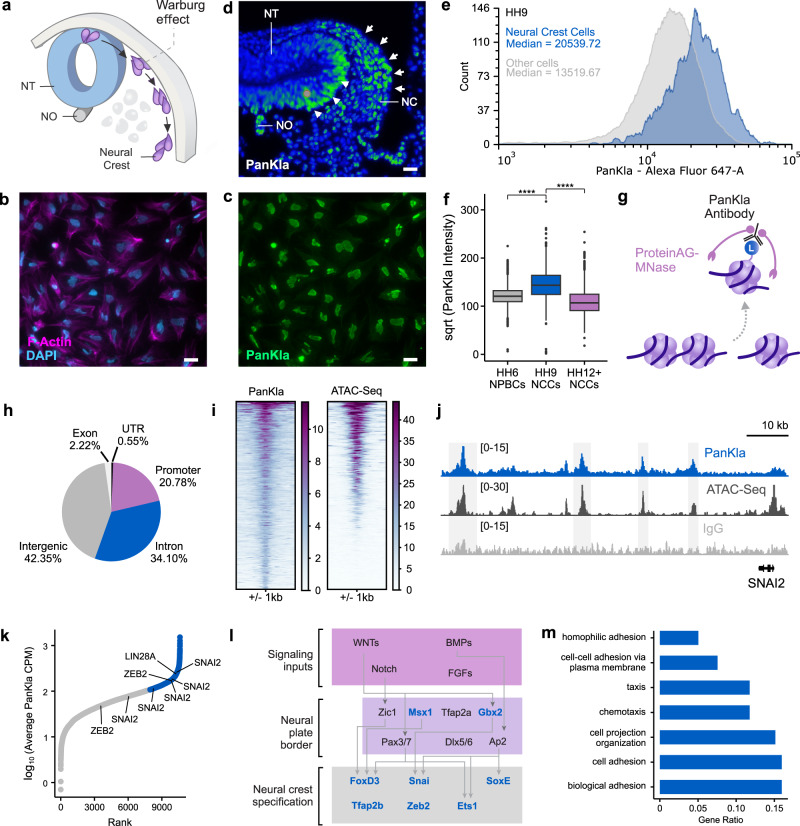
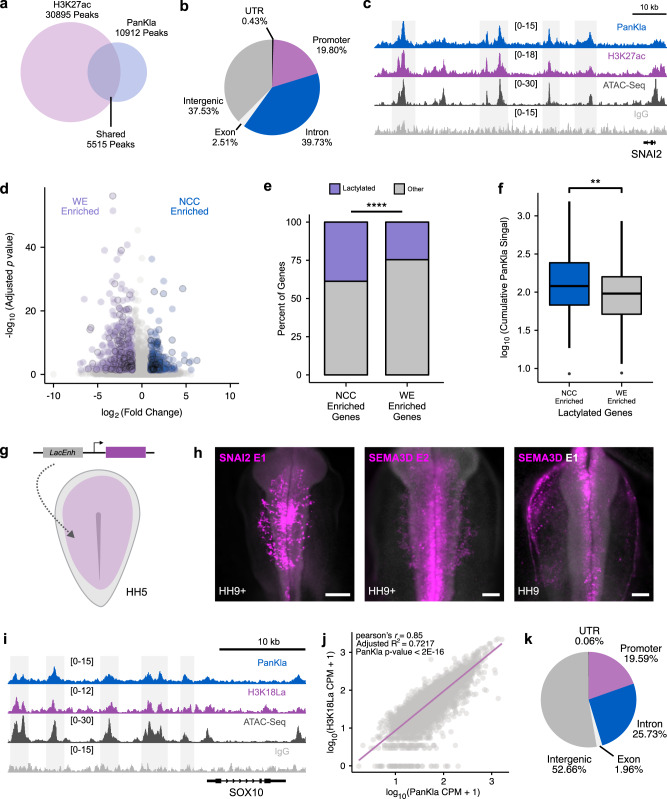
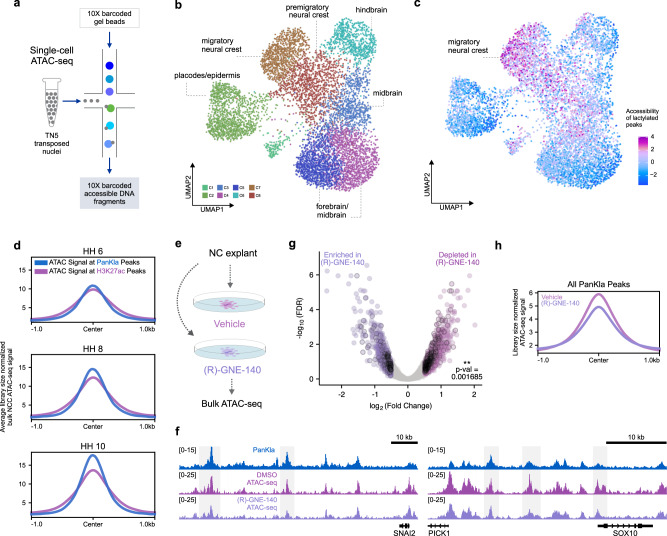
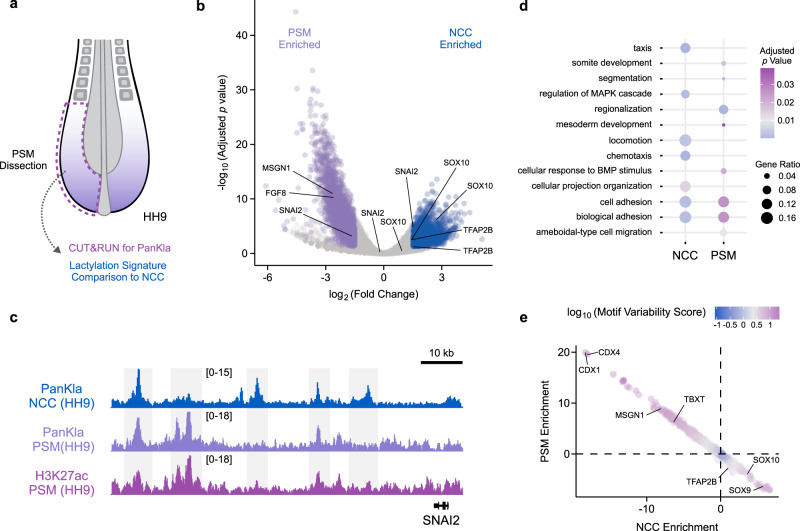
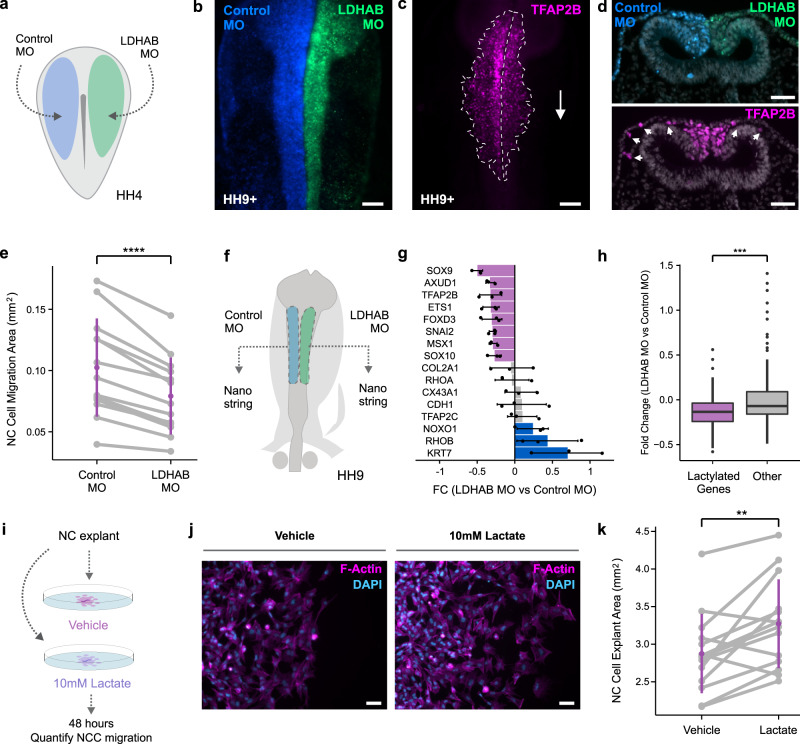
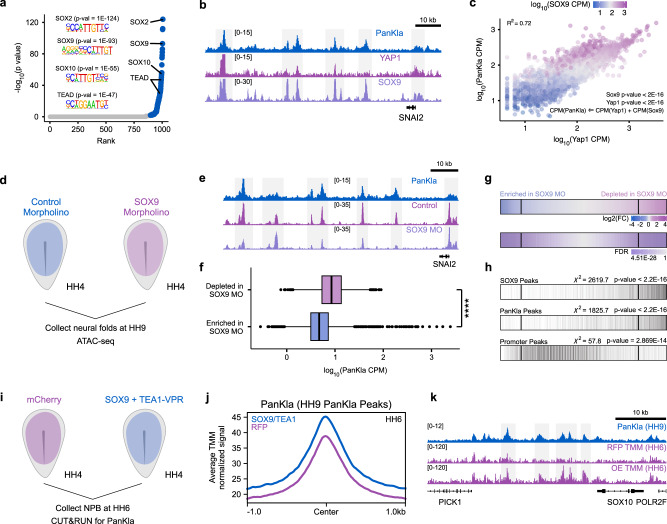
Embryonic cells exhibit diverse metabolic states. Recent studies have demonstrated that metabolic reprogramming drives changes in cell identity by affecting gene expression. However, the connection between cellular metabolism and gene expression remains poorly understood. Here we report that glycolysis-regulated histone lactylation couples the metabolic state of embryonic cells with chromatin organization and gene regulatory network (GRN) activation. We found that lactylation marks genomic regions of glycolytic embryonic tissues, like the neural crest (NC) and pre-somitic mesoderm. Histone lactylation occurs in the loci of NC genes as these cells upregulate glycolysis. This process promotes the accessibility of active enhancers and the deployment of the NC GRN. Reducing the deposition of the mark by targeting LDHA/B leads to the downregulation of NC genes and the impairment of cell migration. The deposition of lactyl-CoA on histones at NC enhancers is supported by a mechanism that involves transcription factors SOX9 and YAP/TEAD. These findings define an epigenetic mechanism that integrates cellular metabolism with the GRNs that orchestrate embryonic development.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
